
Недифференцированные дисплазии соединительной ткани (проект клинических рекомендаций)
Публикуемый проект второго пересмотра клинических рекомендаций по ведению пациентов с недифференцированными дисплазиями соединительной ткани продиктован наличием обоснованных дополнений/замечаний к ранее утвержденным (в 2018 г.) клиническим рекомендациям.
1. ТЕРМИНЫ И ОПРЕДЕЛЕНИЯ
Дисплазия (dysplasia; греч. dys- + plasis формирование, образование; син. дисгенезия) – неправильное развитие тканей и органов независимо от времени и причины их возникновения [1]. Новые и узко направленные профессиональные термины в настоящих клинических рекомендациях не используются.
1.1. Определение
Недифференцированные дисплазии соединительной ткани (НДСТ; код по МКБ-10 – М35.8) – это генетически детерминированные состояния, характеризующиеся дефектами волокнистых структур и основного вещества соединительной ткани, приводящие к нарушению формообразования органов и систем, имеющие прогредиентное течение, определяющие особенности ассоциированной патологии, а также фармакокинетики и фармакодинамики лекарственных средств [2–4].
Комментарии: генетический дефект может проявляться в любом возрасте в соответствии с временными закономерностями генной экспрессии. Реализация генетических детерминант либо в наибольшей степени определяется внешними условиями, как в случае недифференцированных дисплазий соединительной ткани (несиндромных формах дисплазии соединительной ткани, неспецифических нарушений соединительной ткани), либо мало зависит от внешних условий, как в случае наследственных нарушений соединительной ткани (дифференцированной дисплазии соединительной ткани, синдромных форм дисплазии соединительной ткани) [2–7].
1.2. Этиология и патогенез
В основе развития дисплазий соединительной ткани (ДСТ) лежат мутации генов, ответственных за синтез/катаболизм структурных белков соединительной ткани или ферментов, участвующих в этих процессах [2–5] (рис. 1).
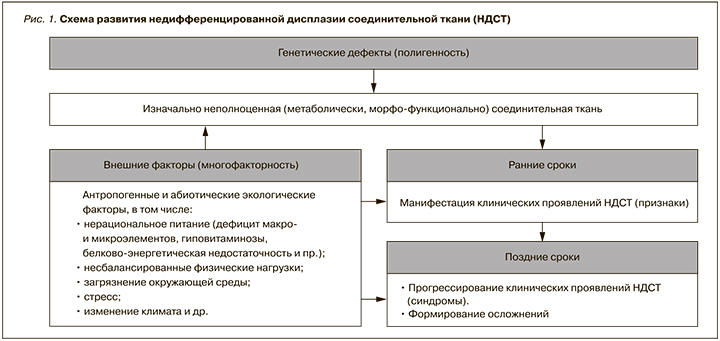
1.3. Эпидемиология
Распространенность НДСТ – 1:5. Отдельные внешние проявления дисморфогенеза соединительной ткани среди молодых – 85,4% [2–4].
Критическим периодом проявлений НДСТ является подростковый возраст, когда объем соединительной ткани увеличивается пропорционально росту и развитию организма. Как правило, у абсолютного большинства пациентов с НДСТ в возрасте старше 35 лет основную проблему составляют осложнения клинических синдромов, определяющие инвалидизацию пациентов и летальные потери в группе [2–4, 8, 9].
1.4. Кодирование по МКБ-10
На территории Российской Федерации диагноз устанавливается по ведущему заболеванию, клиническому синдрому и/или симптому, соответствующему МКБ-10 [10]. Для того, чтобы подчеркнуть полиорганность/полисистемность клинических проявлений НДСТ, в разделе «Диагноз» необходимо указать нозологию, послужившую причиной обращения за медицинской помощью, и далее перечислить все выявленные патологические состояния, присущие НДСТ и имеющие код по МКБ-10, указав при этом НДСТ как фоновое заболевание – «Другие уточненные системные поражения соединительной ткани» (М35.8) [11, 12]. При выявлении макро- и микроэлементозов, гиповитаминозов, также необходимо указать коды МКБ-10, например: E61.2 – «Недостаточность магния»; E59 – «Алиментарная недостаточность селена» и т.п. [10]. Наследственные нарушения соединительной ткани имеют свои собственные коды: синдром Марфана (Q87.4), синдром Элерса–Данло (Q79.6) и т.д. [3, 6].
1.5. Классификация
В практической работе используется Международная классификация болезней 10-го пересмотра (МКБ-10) [10]. В научных исследованиях можно пользоваться классификацией, предложенной Нью-Йоркской ассоциацией кардиологов, с выделением в нозологическую форму соединительнотканной дисплазии сердца, а также каталогом генов и генетических нарушений человека Mendelian Inheritance in Man (MIM), созданном и редактируемом McKusick V.A. et al., в который вошли такие состояния, как MASS syndrome (Mitral valve prolapse, Aortic root diameter at upper limits of normal for body size, Stretch marks of the skin, Skeletal conditions similar to Marfan syndrome, MIM 604308), Mitral valve prolapse, familial (MIM 157700), Mitral valve prolapse, myxomatous 2, 3 (MIM 607829,610840) и ряд других [2–6].
Читайте также: Из чего состоит клеточное строение покровной ткани
1.6. Клиническая картина
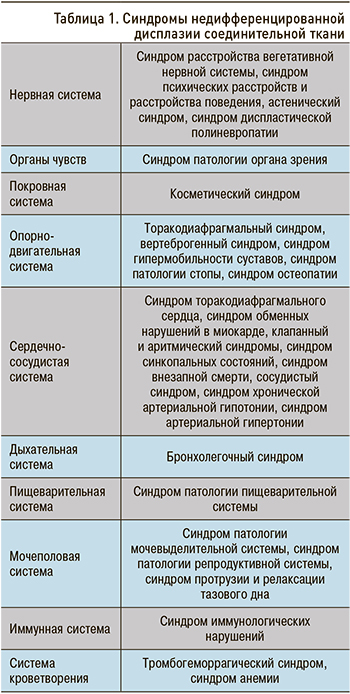
Клинические проявления НДСТ во многом связаны с ведущим клиническим синдромом, затрагивающим ту или иную систему организма (табл. 1). На сегодняшний день выделено 28 синдромов при НДСТ [3].
2. ДИАГНОСТИКА НЕДИФФЕРЕНЦИРОВАННЫХ ДИСПЛАЗИЙ СОЕДИНИТЕЛЬНОЙ ТКАНИ
2.1. Критерии установления диагноза НДСТ
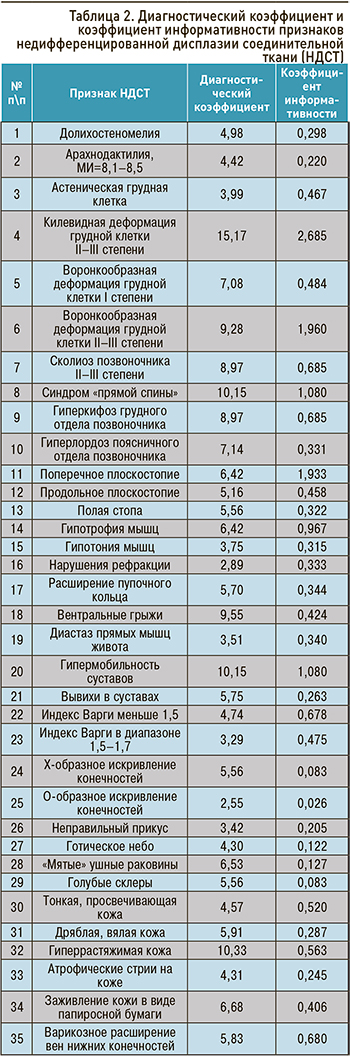
Профессором В.М. Яковлевым и его учениками для верификации диагноза было предложено трактовать клинические проявления пациентов как НДСТ при вовлечении в процесс не менее двух систем (опорно-двигательной, сердечно-сосудистой, бронхолегочной, пищеварительной, нервной, органа зрения и т.д.) с использованием процедуры последовательного распознавания Вальда: на основании диагностических коэффициентов и коэффициентов информативности для определения «диагностического вклада» каждого выявленного у пациента признака (табл. 2, 3) [2–4, 13].
Дисплазия соединительной ткани

Дисплазия соединительной ткани (от др.-греч. δυσ- — приставка, отрицающая положительный смысл слова и πλάσις — «образование, формирование») — системное заболевание соединительной ткани (группа системных заболеваний соединительной ткани) [1] , генетически гетерогенное и клинически полиморфное патологическое состояние (группа генетически гетерогенных и клинически полиморфных патологических состояний) [2] , обусловленное нарушением развития соединительной ткани в эмбриональном и постнатальном периодах. Характеризуется дефектами волокнистых структур и основного вещества соединительной ткани, приводящее к расстройству гомеостаза на тканевом, органном и организменном уровнях в виде различных морфофункциональных нарушений висцеральных и локомоторных органов с прогредиентным течением. [3] Синонимы: cоединительнотканная дисплазия, наследственное нарушение соединительной ткани, врожденная соединительнотканная недостаточность, системное невоспалительное заболевание соединительной ткани, гипермобильный синдром [4] (чаще используют для обозначения несиндромной ДСТ), наследственная коллагенопатия [5] .
Выделяют дифференцированную и недифференцированную дисплазию соединительной ткани. Дифференцированная ДСТ включает в себя синдромы Элерса-Данлоса, Марфана, Стиклера, несовершенного остеогенеза и др. Недифференцированная ДСТ — определяющий вариант ДСТ с клиническими проявлениями, не укладывающимися в структуру наследственных синдромов.
История
Научный и практический интерес к гипермобильности суставов возник еще в конце XIX века, когда были описаны наследственные синдромы, в клинической картине которых гипермобильность суставов являлась одним из ведущих симптомов. Врачи различных специальностей регулярно видели пациентов, у которых имеет место структурное невоспалительное поражение (часто врожденное или проявляющееся в молодом возрасте) отдельных органов или систем.
Группа наследственных заболеваний соединительной ткани и скелета была впервые выделена американским генетиком Mc Kusick в 1955 году. К тому времени она объединяла лишь некоторые нозологические формы: несовершенный остеогенез, синдром Марфана, синдром Элерса-Данло, эластическую псевдоксантому и гаргоилизм.
В 1967 году Кирк (J. H. Kirk), Анселл (B. M. Ansell) и Байватерс (E. G. Bywaters) предложили термин «гипермобильный синдром» для характеристики патологии у пациентов с гиперподвижными суставами и стойкими жалобами со стороны опорно-двигательного аппарата при отсутствии у них признаков какого-либо другого ревматического заболевания. [6] С этого времени началось систематическое изучение указанной патологии в рамках ревматологических синдромов. Термин «гипермобильный синдром» отражал феномен гипермобильности суставов, сочетающийся с дисфункцией опорно-двигательного аппарата (подвывихи, артралгии). [4]
Сегодня, благодаря достижениям генетики были описаны и классифицированы свыше 200 заболеваний соединительной ткани и скелета наследственного характера. [7]
Читайте также: Ткань для зимний костюм цифра


Гипермобильный пястно-фаланговый сустав I пальца

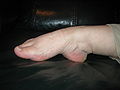
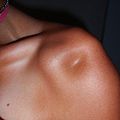



Гипермобильные локтевые суставы
Терминология
В медицине уже более 100 лет известны дифференцированные соединительнотканные дисплазии: синдром Марфана, синдром Элерса-Данлоса, несовершенный остеогенез и др. (они включены в МКБ). Эти заболевания сравнительно редки и имеют четко очерченные диагностические признаки. Однако существовала и группа пациентов, не удовлетворяющая критериям синдромной ДСТ. Очевидная генерализованность вовлечения в процесс соединительнотканных структур привела к широкому использованию обобщающих терминов: «соединительнотканная дисплазия», «врожденная соединительнотканная недостаточность», «недифференцированная наследственная коллагенопатия». В кардиологии распространено понятие соединительнотканных дисплазий сердца, МASS-фенотипа.
Отсутствие общепринятого определения не позволяло сравнивать и обобщать наблюдения разных авторов. Каждая новая публикация очередной раз констатировала «системность» вовлечения в процесс соединительнотканных структур при отдельном интересе авторов к одной из перечисленных в таблице нозологий.
В результате появился международный термин «гипермобильный синдром» (M35.7 в МКБ-10). Он не включил в себя дифференцированные формы соединительнотканной дисплазии. Достоинствами этого термина является выделение генерализованной гипермобильности суставов как наиболее характерного и легко определяемого клинического признака данной группы заболеваний, а отсутствие в определении слова «сустав» ориентирует врача на системные проявления синдрома.
В России чаще всего используют термин «дисплазия соединительной ткани», который включает в себя и синдромные и несиндромные формы. Иногда его также используют для обозначения только недифференцированной соединительнотканной дисплазии, общую группу наследственных коллагенопатий называя при этом «Наследственными нарушениями соединительной ткани».
Этиология
ДСТ морфологически характеризуется изменениями коллагеновых, эластических фибрилл, гликопротеидов, протеогликанов и фибробластов, в основе которых лежат наследуемые мутации генов, кодирующих синтез и пространственную организацию коллагена, структурных белков и белково-углеводных комплексов, а также мутации генов ферментов и кофакторов к ним. Некоторые исследователи, основываясь на выявляемом в 46,6-72,0 % наблюдений при ДСТ дефиците магния в различных субстратах (волосы, эритроциты, ротовая жидкость), допускают патогенетическое значение гипомагниемии. [3]
Классификация
Классификация ДСТ — один из самых дискуссионных научных вопросов. Отсутствие единой, общепринятой классификации ДСТ отражает разногласие мнений исследователей по данной проблеме в целом. ДСТ может классифицироваться с учетом генетического дефекта в периоде синтеза, созревания или распада коллагена. Это перспективный классификационный подход, который дает возможность обосновать генетически дифференцированную диагностику ДСТ, однако на сегодняшний день данный подход ограничен наследственными синдромами ДСТ.
Т. И. Кадурина выделяет MASS-подобный фенотип, марфаноидный и элерсоподобный фенотипы, отмечая, что именно эти три фенотипа являются наиболее частыми формами несиндромной ДСТ. «Марфаноидный фенотип» характеризуется сочетанием «признаков генерализованной дисплазии соединительной ткани с астеническим телосложением, долихостеномелией, арахнодактилией, поражением клапанного аппарата сердца (а порой и аорты), нарушением зрения». При «Элерсоподобном фенотипе» отмечается «сочетание признаков генерализованной дисплазии соединительной ткани с тенденцией к гиперрастяжимости кожи и разной степенью выраженности гипермобильности суставов». «MASS-подобному фенотипу» присущи «признаки генерализованной дисплазии соединительной ткани, ряд нарушений со стороны сердца, скелетные аномалии, а также кожные изменения в виде истончения или наличия участков субатрофии». На основе этой классификации предлагается формулировать диагноз ДСТ. [3] [8]
Клинические проявления
ДСТ — это заболевание с очень разными клиническими проявлениями, от совсем легких в состоянии здоровья до весьма серьёзных и прогностически значимых болезней. Универсальных патологических повреждений соединительной ткани не существует. Каждый дефект у каждого больного в своем роде уникален.