
Липиды накопление которых в тканях происходит при болезни гоше
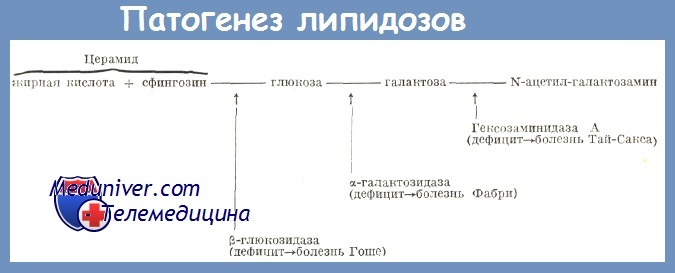
Глюкоцереброзиды состоят из жирной кислоты (вариирующий от одного цереброзида к другому), сфингозина (моноаминированный ненасыщенный диалкоголь с 18 атомами углерода) и глюкозы. Катаболизм происходит путем их деградации на разных уровнях специфическими энзимами, из которых наиболее важными являются глюкозидазы.
Бета-глюкозидаза, вероятно лизозомальный энзим, гидролизирует глюкоцереброзиды в церамид, согласно реакции:
глюкоцереброзиды + вода (с участичем глюкоцереброзидазы) => глюкоза + церамгд.
Сокращение или бездействие энзима блокирует катаболизм глюколипидов в стадии глюкоцереброзидов, за которым следует их накопление в макрофагах и появление клеток Гоше. Глюкоцереброзиды образуются в результате нормального метаболизма и клеточных деструкции.
Главным источником глюколипидов в периферических тканях является лактозидцерамид (главный нейтральный гликолипид гранулоцитов) и липиды в мембране и строме стареющих эритроцитов. Пластинчатый аспект клеток Гоше дан хрусталевидным расположением, в виде тубулярных включений глюколипида, депонированного в цитоплазме. Эти микротрубки появляются в результате слияния вакуолей пиноцитоза, которые каптировали продукты деградации эритроцитов.
Пластинчатое расположение имеет патогномоническое значение для клетки Гоше, по сравнению с пенистым видом, который имеют и клетки Нимана-Пика, и другие ксантоматозные клетки.
Очевидно существует прямая взаимосвязь между уровнем цереброзидов и активностью b-глюкозидазы. Чем ограниченее активность энзима, тем ранее начало болезни и суровее ее эволюция. Однако взаимоотношение между кислотной гиперфосфатаземией и недостаточной деградацией глюкоцереброзидов неизвестно.
Часто встречающиеся в этих клетках аспекты эритрофагоцитоза доказывают, с одной стороны фагоцитарную деятельность клеток Гоше, а с другой стороны подкрепляют эритроцитарное происхождение церебро-зидов и железа, которые они содержат.
Анемия при болезни Гоше имеет в своей основе множество механизмов: дислокация костного мозга, гиперспленизм, гипергемолиз, кровотечения, гемодилюция и т.д. Редко преобладает процесс гемолиза, с явлениями активной регенерации и с макроцитарным аспектом. Нейтропения происходит благодаря гиперспленизму и заполнением костного мозга из-за пролиферации клеток Гоше. Это объясняет восприимчивость больных к инфекциям.
Тромбоцитопения происходит путем механизмов, подобных тем, которые действуют при нейтропении. Оно бывает часто и лежит в основе возникновения геморрагических явлений. Пингвекулы появляются благодаря накоплению клеток Гоше.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Болезнь Гоше: клиническая картина, диагностика, лечение
- КЛЮЧЕВЫЕ СЛОВА: лизосомные болезни накопления, болезнь Гоше, дефицит глюкоцереброзидазы, lysosomal storage diseases, Gaucher disease, glucocerebrosidase deficiency
В повседневной работе практические врачи не всегда имеют дело с распространенными, хорошо известными, социально значимыми заболеваниями. Иногда клинические симптомы выходят за рамки привычного диагностического процесса.
В России о редких заболеваниях и «лекарствах-сиротах» впервые заговорили после введения программы дополнительного лекарственного обеспечения, которая позволила составить реестры больных, определить лекарственную потребность, запланировать необходимые ресурсы.
По данным экспертов, частота заболеваний, относящихся к редким, в разных странах колеблется от одного случая на 1,5 тысячи человек до одного случая на 2,5 тысячи человек. Согласно статистике, в России 1,5 млн пациентов имеют эксклюзивные заболевания, причем 80% из них обусловлены генетической природой.
К редким заболеваниям относится обширный класс наследственных ферментопатий, или лизосомных болезней накопления. Он насчитывает около 40 нозологических единиц и характеризуется генетической гетерогенностью и выраженным клиническим полиморфизмом. Практически все лизосомные болезни накопления имеют прогредиентное течение и без соответствующего лечения приводят к ранней инвалидизации и преждевременной смерти. Лишь немногие формы лизосомных болезней накопления характеризуются близкой к норме продолжительностью жизни.
Наиболее типичными клиническими симптомами лизосомных болезней накопления являются поражение центральной нервной системы, костные нарушения, гепатоспленомегалия, гематологические изменения. Клинические симптомы обусловлены преимущественным накоплением патологического материала в том или ином органе. Нарушение ферментных систем приводит к накоплению соответствующих специфических метаболитов в макрофагальных элементах с последующей инфильтрацией органов и тканей [1–3].
К лизосомным болезням накопления относится и болезнь Гоше – генетическое заболевание, обусловленное дефектом лизосомного фермента бета-D-глюкозидазы. Эта болезнь впервые была описана в 1882 г. французским врачом Филиппом Гоше (Philippe Gaucher). Он описал женщину с анемией, с детства имевшую массивную спленомегалию и страдавшую от тяжелых кровотечений и различных инфекционных осложнений. При аутопсии в ее селезенке были выявлены необычные крупные клетки [4]. И только спустя 80 лет, в 1965 г., Роско Брейди (Roscoe Brady) обнаружил ключевой метаболический дефект, лежавший в основе развития болезни Гоше, – дефицит бета-глюкоцереброзидазы [5, 6].
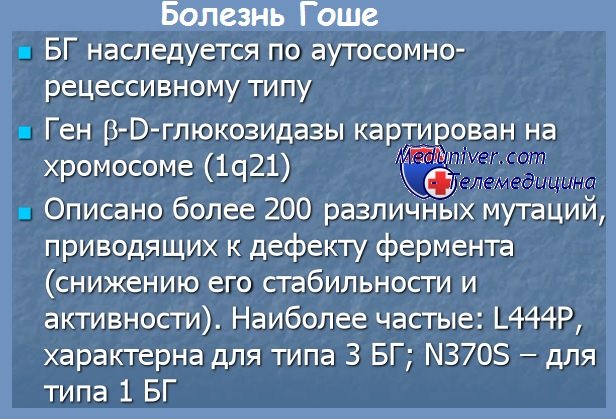
Болезнь Гоше наследуется по аутосомно-рецессивному типу. Ген бета-глюкоцереброзидазы локализуется в регионе q21 на первой хромосоме. Описано около 300 различных мутаций, приводящих к дефекту фермента (снижению его стабильности или активности) и связанных с широким полиморфизмом клинических симптомов заболевания. Наиболее распространенные мутации – N370S, 84GG, L444P, IVS2+1, L444P, D409H, R463C и IVS2+1 [4, 7–10].
Читайте также: Декоративные ткани для мягкой мебели
Вследствие врожденной недостаточности этого фермента в клетках ретикулоэндотелиальной системы, особенно в печени, селезенке и костном мозге, накапливаются неутилизированные липиды.
В силу недостаточной осведомленности врачей диагностика и лечение болезни Гоше затруднительны и сегодня. Как правило, у пациента, не получающего адекватной терапии, полиорганные поражения усложняют клиническую картину заболевания. Такого пациента можно на протяжении многих лет лечить от различных патологий, под маской которых скрывается истинная болезнь [1, 11, 12].
В зависимости от клинической картины выделяют три типа болезни Гоше:
- тип 1 – ненейронопатический (наиболее распространенный);
- тип 2 – инфантильный, или острый нейронопатический;
- тип 3 – подострый нейронопатический.
Типы 2 и 3 являются нейронопатическими, поскольку в патологический процесс вовлекается нервная система.
Болезнь Гоше типа 1 имеет хроническое течение. Возраст манифестации заболевания варьирует от года до 60 лет. Основные клинические симптомы:
- гепатоспленомегалия;
- геморрагический синдром;
- костные боли (костные кризы);
- нарушение подвижности в суставах, обусловленное асептическим некрозом;
- патологические переломы;
- задержка физического и полового развития;
- астенический синдром.
Клиническая картина разнообразна и проявляется прогрессирующим увеличением паренхиматозных органов (печени и селезенки), панцитопенией и патологией трубчатых костей скелета. Характерна своеобразная гиперпигментация кожных покровов в области коленных и локтевых суставов. Спленомегалия – постоянный и наиболее ранний признак болезни Гоше. При пальпации селезенка имеет плотную консистенцию. Гепатомегалия выражена в меньшей степени, чем спленомегалия, и обычно развивается в более поздние сроки или становится выраженной после спленэктомии. При прогрессировании болезни Гоше может развиться портальная гипертензия.
У большинства пациентов наблюдаются хронические боли в костях. При прогрессировании болезни могут наблюдаться патологические переломы, деформация головок бедренных костей, укорочение нижних конечностей и нарушение походки. Характерны костные кризы, сопровождающиеся мучительными болями в костях, гиперемией и болезненностью в области суставов, резким снижением двигательной активности, лихорадкой, ознобом, полной утратой дееспособности. На пике криза отмечаются высокий лейкоцитоз и повышенная скорость оседания эритроцитов. Продолжительность кризов – 12–36 часов в течение 5–15 дней. У большинства больных костные кризы развиваются спонтанно. В 15–40% случаев они могут наблюдаться, в частности, после перенесенной вирусной инфекции. Кризы могут быть обусловлены инфарктами в трубчатых костях (часто в дистальных отделах бедренных, проксимальных отделах большеберцовых и плечевых костей), а могут развиваться без клинически выраженного остеонекроза. Клиническая картина костного криза при болезни Гоше аналогична картине остеомиелита. В тяжелых случаях наблюдается разрушение тел позвонков с последующим компрессионным переломом и образованием углового горба [2, 11, 13–15].
Основные симптомы заболевания при болезни Гоше типа 2 возникают в первые шесть месяцев жизни пациентов. Клинический симптомокомплекс включает признаки поражения нервной системы и внутренних органов. На ранних стадиях заболевания отмечаются мышечная гипотония, задержка и регресс психомоторного развития. По мере прогрессирования болезни появляются спастичность с характерной для этого типа ретракцией шеи, сгибанием конечностей, глазодвигательные нарушения с развитием сходящегося косоглазия, ларингоспазм и дисфагия.
Характерны бульбарные нарушения с частыми аспирациями, приводящие к смерти пациента от апноэ, аспирационной пневмонии или дисфункции дыхательного центра головного мозга.
Тонико-клонические судорожные приступы, как правило, возникают на поздних стадиях болезни и резистентны к назначаемой противосудорожной терапии. Течение заболевания быстро прогрессирующее с летальным исходом на первом-втором году жизни пациента [1, 2, 11, 14, 16].
Особенность клинических проявлений болезни Гоше при этом типе состоит в том, что наряду с поражением паренхиматозных органов (спленомегалия, гепатомегалия) наблюдаются неврологические проявления. Они обычно возникают в возрасте от шести до 15 лет и позже. Характерный симптом – парез мышц, иннервируемых глазодвигательным нервом, который длительное время может быть единственным неврологическим проявлением. Не исключены миоклонии, генерализованные тонико-клонические судороги. Постепенно прогрессируют экстрапирамидная ригидность, снижение интеллекта, тризм, лицевые гримасы, дисфагия, ларингоспазм. Интеллектуальные нарушения варьируют от незначительных изменений до тяжелой деменции. Возможны мозжечковые нарушения, расстройства речи и письма, поведенческие изменения, эпизоды психоза. В большинстве случаев течение заболевания медленно прогрессирующее. Летальный исход наступает при тяжелых поражениях легких и печени.
Продолжительность жизни пациентов при болезни Гоше типа 3 составляет 12–17 лет. Однако описаны случаи выживания до 30–40 лет [8, 11, 15, 17].
Читайте также: Ткань портьерная блэкаут серебро
При подозрении на болезнь Гоше диагностика включает несколько этапов:
- обнаружение характерных клинических признаков заболевания;
- измерение активности бета-D-глюкозидазы в лейкоцитах;
- проведение молекулярно-генетического анализа.
Помимо специфической диагностики применяются рутинные лабораторно-инструментальные методы, позволяющие выявлять определенные изменения.
У большинства больных гиперспленизм проявляется тромбоцитопенией, лейкопенией и анемией.
При ультразвуковом исследовании и компьютерной томографии печени и селезенки определяются очаги как с повышенной, так и с пониженной интенсивностью сигнала. Эти очаги являются зонами ишемии и фиброза из-за повышенной инфильтрации клетками Гоше.
У больных с нейронопатическими типами болезни Гоше изменения, выявляемые при электроэнцефалографии, неспецифичны.
Рентгенограмма позволяет увидеть истончение надкостницы, эндостальную зубчатость и пониженную трабекулярность костной ткани. Дистальные метафизы бедра вздуваются в виде булавы или колбы, которые упоминаются в литературе как колбочки Эрленмейера. У детей отмечается продольное расслоение коркового вещества и линейные костные периостальные отложения на поверхности диафизов трубчатых костей. Тяжесть поражения трубчатых костей различна – от классических деформаций метафизов до тяжелых патологических переломов, очагов литической деструкции и асептических некрозов головок бедренных костей.
Наиболее чувствительными методами выявления сниженной минеральной плотности костной ткани являются денситометрия и магнитно-резонансная томография. Денситометрия при болезни Гоше актуальна как для ранней диагностики системной остеопении, так и для мониторинга эффективности лечения.
Гистологическая картина структур нервной системы при болезни Гоше характеризуется дегенеративными изменениями в тельцах Ниссля, потерей паренхимы в глубоких слоях коры, таламусе, базальных ядрах и мозжечке. При световой микроскопии обнаруживается небольшое количество клеток Гоше вокруг кровеносных сосудов. Ганглиоциты отечны и деформированы. В отличие от других болезней накопления отсутствуют внутриклеточные отложения липидов. В нейронах не определяется отложение глюкоцереброзида.
Молекулярно-генетический анализ – точный метод диагностики болезни, а также гетерозиготных носителей мутаций в гене бета-D-глюкозидазы. Выявление гетерозигот наиболее актуально при пренатальной диагностике и медико-генетическом консультировании.
При проведении дифференциальной диагностики болезни Гоше типа 1 в зависимости от вида манифестации необходимо помнить о разнообразных экзогенных и наследственных заболеваниях, сопровождающихся гепато- и спленомегалией, острыми болями в костях, кровоточивостью (вирусный гепатит, остеомиелит, костный туберкулез, гемофилии, сфинголипидозы). При подозрении на типы 2 и 3 болезни Гоше необходимо исключить все инфантильные формы сфинголипидозов с гепатоспленомегалией (болезнь Ниманна – Пика (Niemann – Pick), типы А и С), GM1-ганглиозидоз, галактосиалидоз, болезнь Вольмана, болезнь Фарбера (атипичные формы), а также врожденную окуломоторную апраксию [1, 2, 4, 5, 15, 16].
Длительное время при лечении болезни Гоше использовалась только симптоматическая и паллиативная терапия (спленэктомия). В 1991 г. N.W. Barton и соавт. впервые применили альглюцеразу (цередазу) – глюкоцереброзидазу, полученную из плаценты человека. На фоне ее применения отмечался регресс патологических изменений. Функции пораженных органов восстанавливались [18]. Это стало прорывом в лечении болезни Гоше. В 1994 г. после проведения клинических исследований к применению была разрешена рекомбинантная глюкоцереброзидаза – имиглюцераза (Церезим®) [6]. В клинических исследованиях доказано, что эффективность Церезима идентична таковой цередазы [19].
Ферментозаместительная терапия – единственный эффективный патогенетический метод лечения болезни Гоше, который купирует основные клинические проявления заболевания, улучшает качество жизни больных и не оказывает выраженных побочных эффектов. Под действием имиглюцеразы происходит гидролиз гликолипида глюкоцереброзида до глюкозы и церамида по обычному пути метаболизма мембранных липидов. Имиглюцераза показана для длительной заместительной ферментотерапии у пациентов с подтвержденным диагнозом болезни Гоше без поражения нервной системы (тип 1) или с хроническим поражением нервной системы (тип 3), у которых имеются клинически значимые не неврологические проявления заболевания [13, 16, 17].
На сегодняшний день в мире свыше 5,5 тысячи пациентов с болезнью Гоше получают специфическую энзимотерапию. Заместительная ферментная терапия – процедура дорогостоящая. В развитых странах мира такое лечение поддерживается специальными государственными программами, и пациенты получают его бесплатно [6]. В России бесплатная заместительная ферментная терапия болезни Гоше стала доступна с 2006 г. [17].
Показаниями к началу заместительной терапии служат детский возраст, цитопения, клинические и рентгенологические признаки поражения костей, значительная сплено- и гепатомегалия, симптомы поражения легких и других органов. Первоначальная доза имиглюцеразы для взрослых пациентов с болезнью Гоше типа 1 составляет 30 ЕД/кг, вводится внутривенно капельно медленно один раз в 14 дней. В отдельных случаях (поражение костей, тяжелый остеопороз с повторными патологическими переломами трубчатых костей, поражение легких с развитием легочной гипертензии или гепатопульмонарного синдрома) доза может быть повышена до 60 ЕД/кг на одно введение [16, 17, 20–22]. У детей с болезнью Гоше типа 1, протекающей с поражением трубчатых костей скелета (костные кризы, патологические переломы, очаги литической деструкции, асептический некроз головок бедренных костей), имиглюцераза назначается по 60 ЕД/кг один раз в 14 дней. При типе 3 доза повышается до 120 ЕД/кг и вводится один раз в 14 дней.
Читайте также: Химчистка для деликатных тканей
Постоянная ферментная заместительная терапия приводит к регрессу патологических изменений пораженных органов и восстановлению их функций. Кроме того, на фоне терапии отмечается значительное улучшение показателей физического развития, снижается количество интеркуррентных заболеваний, характерных для болезни Гоше.
После достижения поставленных целей лечения у взрослых доза имиглюцеразы постепенно снижается до поддерживающей – 15–30 ЕД/кг в месяц [16, 17].
У 6–7% больных развивается непереносимость энзимотерапии, которую в большинстве случаев удается купировать предварительным введением антигистаминных препаратов и/или глюкокортикостероидов.
Имиглюцераза применяется в клинической практике свыше 20 лет. За это время был получен опыт применения Церезима у взрослых пациентов с болезнью Гоше, детей, беременных, а также у пациентов с типом 3 болезни. Опубликованы результаты ферментозаместительной терапии у пациентов, в течение десяти лет получавших альглюцеразу (цередазу) или имиглюцеразу (Церезим®). Продемонстрировано значительное устойчивое улучшение состояния пациентов с болезнью Гоше, которое оценивалось по таким параметрам, как уровень гемоглобина и тромбоцитов, объем печени и селезенки (у неспленэктомизированных больных), наличие костных болей и костных кризов [23].
Кроме применения имиглюцеразы есть еще один способ лечения: уменьшение выработки субстрата (собственно глюкоцереброзида) путем ингибирования фермента глюкоцерамидсинтетазы. Таким механизмом действия обладает препарат миглустат (Завеска®), принимаемый перорально. На фоне его приема наблюдаются уменьшение размеров печени и селезенки, снижение явлений гиперспленизма (повышение гемоглобина и количества тромбоцитов) [24, 25].
В России проводятся клинические исследования перорального препарата субстратредукционной терапии. Новая таблетированная форма препарата позволит существенно повысить качество жизни пациентов с болезнью Гоше независимо от места проживания, поскольку периодического посещения медицинского центра для внутривенных инфузий не потребуется [13].
Cейчас проводятся исследования в области генной инженерии. Предполагается, что генная терапия будет связана с введением нормальных генов глюкоцереброзидазы в клетки больного. Эти клетки впоследствии должны производить достаточное количество собственной глюкоцереброзидазы, что по сути будет означать излечение от болезни Гоше [15].
Комплексная терапия проявлений остеопороза направлена на замедление и прекращение потери костной массы, повышение прочности кости, предотвращение переломов костей и включает назначение бисфосфонатов, альфакальцидола, солей кальция, Остеогенона.
Симптоматическое лечение скелетных осложнений при болезни Гоше типа 1 предполагает применение анальгетиков во время костных кризов, антибактериальной терапии. При хирургических вмешательствах существует повышенный риск кровотечения и инфицирования [11, 13, 20, 22, 26]. Методы эффективной терапии для типа 2 не описаны.
Оценка эффективности лечения
Контроль течения заболевания у детей на фоне терапии проводится в соответствии с рекомендациями по минимально необходимому мониторингу состояния больных при болезни Гоше типа 1, разработанных Международной объединенной группой по изучению болезни Гоше (International Collaborative Gaucher Group – ICGG). При этом контроль анализов крови необходимо проводить один раз в три месяца, размеров паренхиматозных органов (по данным ультразвукового исследования) – один раз в шесть месяцев. Такой контроль следует осуществлять и при изменении дозировки или значительных клинических осложнениях. Контроль состояния костной ткани проводится один раз в год. Особую роль при проведении мониторинга в процессе патогенетического лечения играет определение хитотриозидазы, которая синтезируется макрофагами и является маркерным ферментом лизосомных болезней. Активность хитотриозидазы на фоне ферментозаместительной терапии болезни Гоше определяется один раз в четыре месяца [13, 16, 17].
Прогноз болезни Гоше при типах 1 и 3 зависит от выраженности клинических проявлений. Назначение патогенетической терапии на ранних стадиях заболевания определяет благоприятный прогноз и улучшает качество жизни пациентов, предотвращая их инвалидизацию.
При болезни Гоше типа 2 прогноз крайне неблагоприятен (летальный исход на первом-втором году жизни) [2, 11, 16, 27].
Благодаря успехам медицины эффективное лечение болезни Гоше и других наследственных заболеваний становится реальностью. Применение современных лекарственных средств позволяет уменьшить боль и страдания людей, дает надежду тяжелым пациентам на полноценную жизнь.
- Свежие записи
- Балкон в многоквартирном доме: является ли он общедомовым имуществом?
- Штраф за остекление балкона в 2022: что это и как избежать наказания
- Штраф за мусор с балкона: сколько заплатить за выбрасывание окурков
- Оформление балконного окна: выбираем шторы из органзы
- Как выбрать идеальные шторы для маленькой кухни с балконом