
Аммиак непрерывно образуется во всех органах и тканях организма. Наиболее активными его продуцентами в кровь являются нервная ткань, печень, кишечник, мышцы. Основными его источниками:
• неокислительное дезаминирование некоторых аминокислот(серина, треонина, гистидина) в печени;
• дезаминирование амидов глутаминовой и аспарагиновой кислот– в печени и поч-ках;
• катаболизм биогенных аминов– во всех ткани, в наибольшей степени в нервной ткани;
• жизнедеятельность бактерий толстого кишечника
• распад пуриновых и пиримидиновых оснований– во всех тканях.
• синтез глутаминовой к-ы– происходит практически во всех тканях, но имеет небольшое значе-ние
• синтез глутамина– главный способ уборки. Наиболее активно происходит в нервной и мышечной
тканях, в почках, сетчатке глаза, печени. Реакция протекает в митохондриях;
• синтез аспарагина– является вто-ростепенным способом уборки ам-миака, энергетически невыгоден,
т.к. тратится2 макроэргические связи;
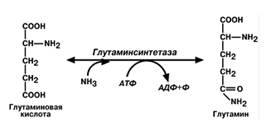
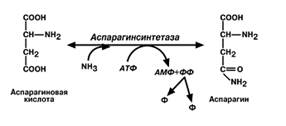
Транспортными формами аммиака являются глутамин, аланин, некоторое кол-во аммиака находится в крови в свобвиде. Большая часть глутамина поступает от мышц и мозга, аланин-от мышц и ст кишечника. Роль глутамина
Образование большого количества глутамина при обезвреживании аммиака обеспечивает высокие концентрации этого вещества в крови(0,5-0,7 ммоль/л). Так как глутамин проникает через клеточные мембраны путем облегченной диффузии, то он легко попадает не только в гепатоциты, но и в другие клетки, где есть потребность в аминогруппах. Азот, переносимый глутамином, используется клетками для синтеза пуринового и пиримидинового колец, ГМФ, аспарагина.
Целевыми органами для транспорта аммиака являются печень, почки и кишечник.
• в кишечнике часть глутамина дезаминируется, образованный аммиак выделяется в просвет кишечника(не более5%), часть поступает в печень, около90% – в мочу;
• в печени происходит синтез мочевины;
• в почках идет образование аммонийных солей.
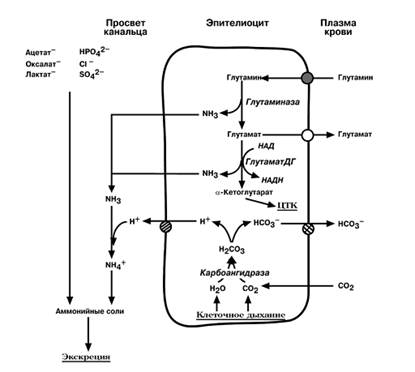
СИНТЕЗ АММОНИЙНЫХ СОЛЕЙ
Часть глутамина крови, не задержавшаяся в печени, достигает почек. В клетках дистальных почечных канальцев имеется фермент глутаминаза, гидролизующая амидную группу в виде аммиака, с образованием глутамата. Глутамат в свою очередь, дезаминируется глутаматдегидрогеназой. Выделяемый аммиак диффундируетв просвет канальца, где соединяется с ионом Н+, образуя ионы аммонияNH4+. Онисвязываются с неорганическими(Cl– иSO42-), или с органическими анионами(ук-сусной, щавелевой, молочной кислот). При ацидозе(закислении крови) необходимость выведения ионов Н+ вызывает увеличение синтеза фермента и возрастание экскреции солей аммония. При алкалозе(защелачивании крови) количество глутаминазы снижается и ионы Н+ сберегаются в организме.
Токсичность аммиака обусловлена:
1. При синтезе глутамата происходит отток α-кетоглутарата из ЦТК, при этом понижается образование энергии АТФ и ухудшается деятельность клеток;
2. Ионы аммонияNH4+ вызывают защелачивание плазмы крови. При этом повышается сродство гемоглобина к кислороду(эффект Бора), гемоглобин не отдает кислород в капиллярах, в результате наступает гипоксия клеток;
3. Накопление свободного ионаNH4+в цитозоле влияет на работу внутриклеточных ферментов и ионных каналов для Na+ и K+;
4. Глутамин, являясь осмотически активным веществом, приводит к задержке воды в клетках и их набуханию, что вызывает отек тканей. В случае нервной ткани это может вызвать отек мозга
5. Использование глутамата для нейтрализации аммиака вызывает снижение синтеза ГАМК, тормозного медиатора нервной системы.
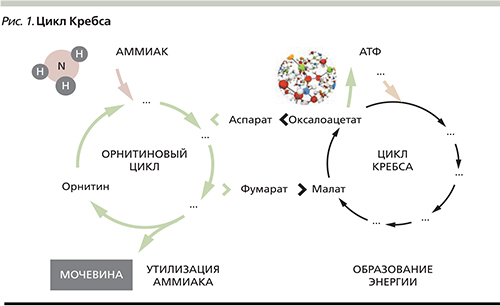
Орнитиновый цикл. Мочевина- полный амид угольной кислоты — содержит 2 атома азота.
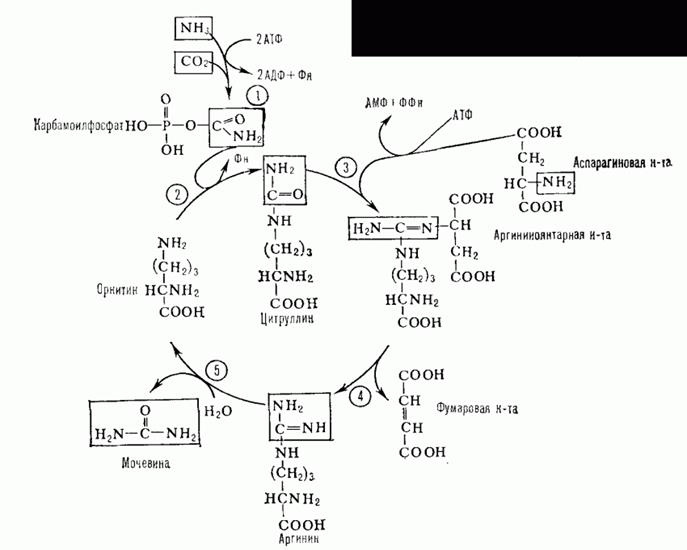
Суммарное уравнение синтеза мочевины:СО2 + NH3 + Аспартат + 3 АТФ + 2 Н2О → Мочевина + Фумарат + 2 (АДФ + Н3Р04 + АМФ + H4P2O7.
В печеночные клетки аммиак попадает в виде глутамина, глутаминовой кислоты, аланина, в свободном виде. Кроме этого, при метаболизме он образуется в большом количестве и в самих гепатоцитах. В печени весь аммиак используется для синтеза мочевины. Увеличение синтеза мочевины наблюдается при голодание, избыточные физические нагрузки, сахарный диабет или избыточном белковом питании.
Синтез мочевины начинается в митохондриях(первая и вторая реакции), оставшиеся три реакции идут в цитозоле. Для переноса цитруллина и орнитина через митохондриальную мембрану существуют специальные переносчики.
В образовании одной молекулы мочевины участвует1 молекулаNH3, 1 молекулаCO2, аминогруппа1 молекулы аспарагиновой кислоты, затрачивается4 макроэргических связи трех молекул АТФ.
В биологических жидкостях М. определяют с помощью газометрических методов, прямых фотометрических методов, основанных на реакции с образованием эквимолекулярных количеств окрашенных продуктов, а также ферментативных методов с использованием уреазы. Газометрические методы основаны на окислении М. гипобромитом натрия в щелочной среде NH2—СО—NH2 + 3NaBrO → N2 + CO2 + 3NaBr + 2H2O. Объем газообразного азота измеряют с помощью аппарата Бородина. Однако этот метод обладает низкой специфичностью и точностью. Из фотометрических наиболее распространены методы, основанные на реакции М. диацетилмонооксимом (реакция Ферона)в присутствии тиосемикарбазида и солей железа в кислой среде. Другим унифицированным методом определения М. является уреазный метод: NH2—СО—NH2 → уреаза NH3 +CO2. Выделившийся аммиак образует с гипохлоритом натрия и фенолом индофенол, имеющий синий цвет. Интенсивность окраски пропорциональна содержанию М. в исследуемой пробе. Уреазная реакция высокоспецифична, для исследования берут лишь 20 мкл сыворотки крови, разведенной в соотношении 1: 9 раствором NaCI. Иногда вместо фенола используют салицилат натрия; сыворотку крови разводят следующим образом: к 10 мкл сыворотки крови добавляют 0,1 мл NaCI . Ферментативная реакция протекает при 37° в течение 3 1 /2 мин
Читайте также: Брюки из ткани шотландка
Основные источники аммиака в тканях
Кафедра поликлинической терапии ФГБОУ ВО МГМСУ им. А.И. Евдокимова Минздрава России, Москва
Основным источником образования аммиака в организме человека является азот пищевого белка, образующийся в ходе реакций дезаминирования аминокислот в печени. Дополнительными источниками образования аммиака являются:
- Разложение мочевины и белка уреазаположительной микрофлорой желудочно-кишечного тракта (ЖКТ).
- Образование аммиака в мышечной ткани при физической нагрузке.
- Распад глутамина в тонкой кишке.
- Абсорбция аммиака в почках при гипокалиемии и/или алкалозе.
Выделение мочевины осуществляется преимущественно через почки (около 80%), примерно 20% мочевины повторно поступают в ЖКТ, где вновь разлагается уреазаположительными бактериями до аммиака.
Детоксикация аммиака в организме осуществляется преимущественно в митохондриях перипортальных гепатоцитов за счет связывания в орнитиновом цикле с аминокислотами и образованием нетоксичной мочевины (рис. 1). Частично детоксикация аммиака происходит в мышечной ткани в процессе синтеза глутамина при участии фермента глутаминсинтетазы. Эта реакция с меньшей интенсивностью протекает также в астроцитах головного мозга и перивенозных гепатоцитах печени. Образующийся в результате этих превращений глутамин нетоксичен и выделяется с мочой.
Таким образом, являясь основным источником аммиака, печень в то же время служит главным местом его обезвреживания. Именно поэтому гипераммониемия развивается в организме человека, прежде всего при хронических заболеваниях печени (ХЗП). К причинам этого относятся снижение активности орнитинового цикла и глутаминсинтетазной реакции при печеночно-клеточной недостаточности и порто-системное шунтирование при развитии и прогрессировании портальной гипертензии.
Значительно реже в рутинной клинической практике встречаются генетически детерминированные ферментопатии, которые сопровождаются повышением концентрации аммиака в сыворотке крови. В зависимости от дефицита или дефекта того или иного фермента выделяют несколько видов генетических заболеваний: гипераммониемию типа I (в основе – дефект карбамоилфосфатсинтетазы I), гипераммониемию типа II (в основе – дефект орнитинкарбамоилтрансферазы), цитруллинемию (в основе – дефект аргининосукцинатсинтетазы), аргининосукцинатурию (в основе – дефект аргининосукцинатлиазы), гипераргининемию (в основе – дефицит аргиназы).
Известно, что аммиак является одним из важнейших нейротоксических метаболитов в организме человека. В клинической практике наиболее частым проявлением гипераммониемии является печеночная энцефалопатия (ПЭ) при патологии печени, представляющая собой спектр нервно-психических расстройств на фоне острой или хронической печеночно-клеточной недостаточности и/или портосистемном шунтировании крови. В основе патогенеза ПЭ лежит дисбаланс аминокислот в головном мозге, приводящий к отеку астроглии и нарушениям ее функций, таких как изменения постсинаптических рецепторов и процессов нейротрансмиссии, нарушение проницаемости гематоэнцефалического барьера, снижение энергетического обеспечения нейронов. Степень клинических проявлений напрямую коррелирует с уровнем аммиака в сыворотке крови. В зависимости от выраженности нарушений деятельности головного мозга выделяют четыре степени тяжести печеночной энцефалопатии (от минимальной до комы).
В последние годы в связи с совершенствованием диагностических методик актуально отделение клинически выраженных стадий ПЭ (дезориентация, атаксия, кома) от стадии с минимально выраженными проявлениями (латентная ПЭ). Такую ПЭ можно выявить, используя специальные опросники или метод вызванных потенциалов головного мозга. При этом выявляются когнитивные и психомоторные расстройства, такие как трудности с принятием решений, снижение скорости психомоторных реакций и др. К клиническим симптомам латентной ПЭ относятся повышенная утомляемость, слабость, раздражительность, инверсия сна (сонливость днем и бессонница ночью), нарушения речи, изменения почерка, рассеянность за рулем и при выполнении работы, требующей повышенной концентрации внимания, тремор, снижение мышечных рефлексов.
К сожалению, результатом таких нарушений могут стать серьезные дорожно-транспортные происшествия с тяжелыми последствиями [1, 2]. В связи с этим выявление минимальной ПЭ имеет большое значение для работников многих профессий: водителей автотранспорта, операторов на автоматизированном оборудовании и др.
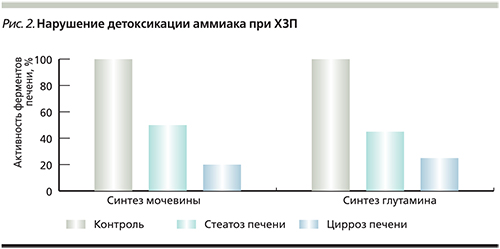
В исследованиях показано, что у больных стеатозом печени при отсутствии клинических признаков воспаления и печеночно-клеточной недостаточности уже имеется выраженное снижение детоксикации аммиака по обоим путям (уменьшается синтез и мочевины, и глутамина) за счет выраженного снижения активности соответствующих ферментов в печени (рис. 2).
Читайте также: Кусочки ткани при менструации
В 2016 г. ученые из Великобритании получили новые научные данные, свидетельствующие о том, что гипераммониемия активирует звездчатые клетки печени (ЗКП) и как следствие – может приводить к усиленному коллагенообразованию и прогрессирующему фиброзированию [3].
Известно, что ЗКП являются основными профиброгенными клетками органа. Хроническое повреждение печени под влиянием различных этиологических факторов (алкоголь, вирусная инфекция, лекарства, холестаз и др.) способствует их активации и дифференцировке в миофибробластоподобные клетки, которые приобретают сократительные, провоспалительные и фиброгенетические свойства. При этом ЗКП пролиферируют, из них исчезают капли жира, увеличивается шероховатая эндоплазматическая сеть, в них появляется специфический белок гладких мышц (α-актин), увеличивается количество рецепторов к цитокинам, стимулирующим пролиферацию и фиброгенез.
К факторам, активирующим ЗКП, относятся трансформирующий фактор роста (TGF-β1 – Transforming growth factor beta), тромбоцитарный фактор роста (PDGF – Platelet-derived growth factor), фактор роста фибробластов, интерлейкин-1 (ИЛ-1), эпидермальный фактор роста, фактор некроза опухоли α. Среди всех факторов роста TGF-β1 позиционируется как ключевой медиатор в фиброгенезе у человека. Различные способы, воздействующие на синтез этого фактора или на сигнальные пути, которые реализуются с участием этого фактора, значимо снижают фиброз в экспериментальных моделях [4].
Активированные ЗКП мигрируют и аккумулируются в месте поражения ткани печени, при этом секретируя большое количество внеклеточного матрикса и одновременно регулируя деградацию этих молекул на уровнях транскрипции и посттранскрипции. Повышение содержания информационной коллагеновой РНК является опосредующим фактором, повышающим синтез коллагена активированными ЗКП. В этих клетках посттранскрипционная регуляция коллагена осуществляется путем последовательности 3-го нетранслируемого региона РНК-связующего протеина aСР2, равно как и посредством структуры в 5-м окончании коллагена информационной РНК. Кроме этого ЗКП экспрессируют большое количество нейроэндокринных маркеров (реелин, нестин, нейротрофины, синаптофизин и глиально-фибриллярные кислотные протеины), а также несут рецепторы нейротрансмиттеров, выделяют провоспалительные цитокины, нейрофильный и моноцитарный хемоаттрактаны, которые усиливают воспалительную реакцию в пораженной печени [5].
Фиброз печени является основным, этиологически независимым путем прогрессирования хронических диффузных заболеваний печени вплоть до цирроза. Печеночный фиброз ассоциируется с изменением количества и качественного состава экстрацеллюлярного коллагенового матрикса (ЭКМ). При выраженных стадиях фиброза печень содержит приблизительно в 6 раз больше ЭКМ, чем в норме, а в его составе определяются коллагены (1-го, 3 и 4-го типов), фибронектин, ундулин, эластин, ламинин, гиалуронан и протеогликаны. Снижение скорости резорбции ЭКМ и выведение молекул металлопротеиназ являются в основном следствием перевысвобождения их специфических ингибиторов (TIMPs – tissue inhibitors of metalloproteinases). Результатом превалирования процессов образования внеклеточного матрикса над его разрушением является формирование фиброзного рубца, при этом фиброз на ранних стадиях развития – процесс обратимый, а цирроз с характерными сшивками между коллагеновыми волокнами и узлами регенерации необратим. Прогрессирующее накопление и отложение внеклеточного матрикса в пространстве Диссе приводят к исчезновению фенестров эндотелия, капилляризации и стенозированию синусоидов с постепенным развитием портальной гипертензии [6–8].
Таким образом, в прогрессировании ХЗП главенствующую роль играют повреждения и ишемия гепатоцитов, которые запускают регенераторные процессы воспаления и коллагенообразования. В свою очередь повышенное коллагенообразование из-за избыточного отложения внеклеточного матрикса и нарушения портального кровотока также приводит к ишемии и некрозу гепатоцитов. Таким образом, можно говорить о «circulus vitiosus» (порочном круге) в прогрессировании заболеваний печени.
Почему ученые из Великобритании считают, что гипераммониемия способна выступить важным фактором в этом «circulus vitiosus»? Такие выводы были сделаны на основе результатов собственного исследования, состоявшего из двух частей: in vitro и in vivo. Первичные ЗКП, полученные из печени здоровых доноров (hHSCs – human hematopoietic stem cells), были посеяны (плотность – 26×103/см2) в исходных, богатых сывороткой условиях (CM – полная среда) на 24 часа с последующим удалением сыворотки на следующие 24 часа (SFM – Serum-free medium). Экзогенный глутамин был удален из культуральной среды во избежание искажения результатов эксперимента.
In vitro показано, что аммиак дозозависимо снижает клеточную пролиферацию и метаболизм в первичных человеческих ЗКП, но не вызывает гибели клеток.
Длительная обработка клеток аммиаком (до 72 часов) индуцирует развитие и прогрессирование эндоретикулярного стресса в hHSCs, что проявлялось выраженным перинуклеарным накоплением красителя (ER-Tracker™ Red) и появлением цитоплазматических вакуолей по мере роста концентрации аммиака. Использование Image IT™-набора для определения зеленых активных форм кислорода позволило исследователям выявить аммиак-дозозависимое образование активных радикалов кислорода (ROS – Reactive oxygen species) в hHSCs. Таким образом, установлено, что увеличение концентрации аммиака и время его воздействия напрямую влияют на уровни мРНК экспрессии маркеров стресса [3].
Читайте также: Функции тканей для человека
Исследование также показало, что при повышенных концентрациях аммиака hHSCs приобретают выраженный профиброгенный и провоспалительный потенциал, что подтверждается результатами исследования in vitro:
- аммиак значительно увеличивает экспрессию белка α-SMA, являющегося активатором ЗКП, а при уровне аммиака 300 мкМ увеличивается синтез виментина – важного промежуточного филамента;
- возрастание концентраций аммиака сопровождается увеличением уровней миозина IIa (играющего ключевую роль в сокращении HSC), миозина IIb (важный фактор активации HSC); экспрессией p38 MAPK, ростом уровней PDGF-Rβ и коллагена I типа;
- повышение уровней аммиака индуцирует сильный и значимый рост мРНК экспрессии металлопротеиназ-2, в то время как мРНК-экспрессия TIMP1 снижается;
- при обработке hHSC аммиаком в концентрации 300 мкМ на протяжении 72 часов мРНК значительно возрастает экспрессия провоспалительного ИЛ-1β;
- аммиак в дозах 50 и 100 мкМ значительно увеличивает в hHSC мРНК- экспрессию ИЛ-6, но не изменяет мРНК-экспрессию ИЛ-8.
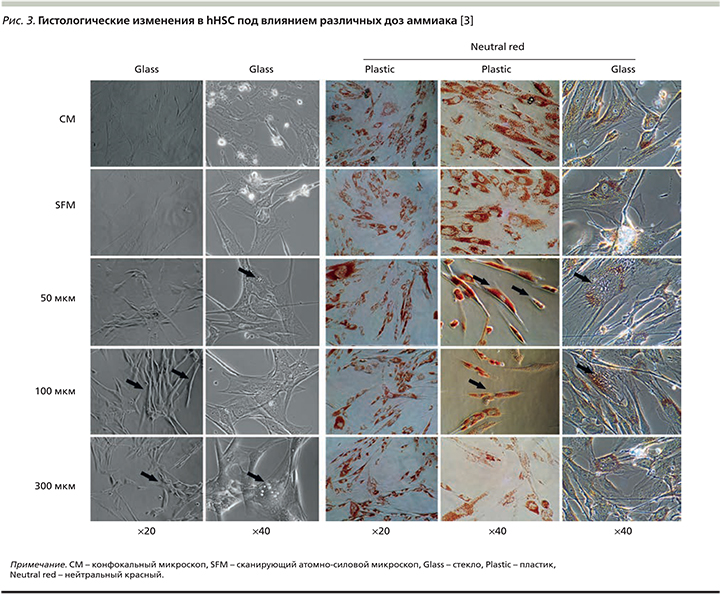
При гистологическом изучении клеток установлено, что аммиак дозозависимо вызывает серьезные морфологические изменения. Так, при световой микроскопии и в тесте жизнеспособности с нейтральным красным (20х, 40х) наблюдали превращение миофибробластоподобных клеток в веретеноподобные фибробласты под действием повышенных концентраций аммиака (рис. 3). Электронная микроскопия показала изменения структуры цитоскелета клеток с образованием цитоплазматических вакуолей при гипераммониемии, однако эти изменения быстро регрессировали после помещения клеток в безаммиачную среду [3].
Вторая часть исследования была проведена на самцах крыс, которые были разделены на три группы: в двух группах проведено перевязывание общего желчного протока (BDL – экспериментальная модель холестаза) с целью формирования экспериментального повреждения печени, при этом крысам одной из групп вводился орнитин (ОР), второй – физиологический раствор. Третья группа животных была контрольной, без повреждения печени.
Анализ результатов опыта показал, что концентрации аммиака были значительно увеличены в плазме крыс с BDL по сравнению с контролем (182±12,8 против 62,51±6,2 мкМ; p
1. Bajaj J.S., Pinkerton S.D., Sanyal A.J., Heuman D.M. Diagnosis and treatment of minimal hepatic encephalopathy to prevent motor vehicle accidents: a cost-effectiveness analysis. Hepatology. 2012;55(4):1164–71.
2. Богомолов П.О., Буеверов А.О., Уварова О.В., Мациевич М.В. Гипераммониемия у пациентов с заболеваниями печени на доцирротической стадии: возможно ли это? Клин. перспективы гастроэнтерол., гепатол. 2013;5:3–8.
3. Джалан Р., Де Чиара Ф., Баласубраманиян В., Андреола Ф., Кхетан В., Малаго М., Пинзани М., Мукерджи Р.П., Ромбоутс К. Аммиак приводит к патологическим изменениям в звездчатых клетках печени, и является целью при лечении портальной гипертензии. Журн. гепатологии. 2016;64:823–33.
4. Gressner A.M., Weiskirchen R., Breitkopf K., Dooley S. Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 2002;7:d793–d807.
5. Lindquist J.N., Parsons C.J., Stefanovic B., Brenner D.A. Regulation of alpha1(I) collagen messenger RNA decay by interactions with alphaCP at the 3?-untranslated region. J. Biol. Chem. 2004;279:23822–29.
6. Benyon R.C., Iredale J.P. Is liver fibrosis reversible? Gut. 2000;46:443–46.
7. Arthur M.J. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:G245–G249.
8. Arthur M.J. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002;122:1525–28.
9. Агеева Е.А., Алексеенко С.А. Опыт применения пероральной формы препарата «L-орнитин-L-аспартат» при гипераммониемии у больных с хроническими заболеваниями печени на доцирротической стадии. Клин. перспективы гастроэнтерологии, гепатологии. 2015;6:24–6.
10. Бурков С.Г., Арутюнов А.Г., Годунова С.А., Гурова Н.Ю., Егорова Н.В., Должикова Т.А., Шиковная Ю.Н. Эффективность гранул L-орнитин-L-аспартата в лечении неалкогольной жировой болезни печени. Consilium Medicum. 2010;12(8):43–7.
11. Осипенко М.Ф., Редькина А.В., Бикбулатова Е.А., Моисеенко Е.Е., Скалинская М.А., Казакова Е.А. Оценка L-орнитин-Lаспартата (Гепа-Мерц) в комплексном лечении неалкогольного стеатогепатита. Consilium Medicum. Прил. Гастроэнтерология. 2010;1:35–8.
12. Грюнграйфф К., Ламберт-Бауманн Й. Эффективность гранул L-орнитин-L-аспартата при лечении хронических заболеваний печени. Сучасна гастро-ентерологія. 2008;2:59–67.
13. Ермолов С.Ю., Шабров А.В., Ермолова Т.В. и др. Новые подходы к диагностике и коррекции портопеченочной гемодинамики. Эксперим. и клин. гастроэнтерология. 2007;4:13–6.
14. Ермолова Т.В., Яковлева Д.М. Эффективность применения L-орнитина-L-аспартата у больных стеатогепатитом. Соврем. гастроэнтерология и гепатология. 2012;1:22–6.
- Свежие записи
- Балкон в многоквартирном доме: является ли он общедомовым имуществом?
- Штраф за остекление балкона в 2022: что это и как избежать наказания
- Штраф за мусор с балкона: сколько заплатить за выбрасывание окурков
- Оформление балконного окна: выбираем шторы из органзы
- Как выбрать идеальные шторы для маленькой кухни с балконом