
Тема 5. Обмен нуклеопротеинов. Биосинтез и распад пуриновых и пиримидиновых нуклеотидов.
Практическая значимость темы. Нуклеотиды — одни из наиболее важных компонентов клетки, принимающие участие во многих биохимических процессах. Они являются структурными единицами ДНК и РНК, участвуют в активации промежуточных продуктов реакций биосинтеза (УДФ-глюкоза, ЦДФ-холин), входят в состав коферментов (НАД, ФАД, КоА-SH, АТФ), служат аллостерическими регуляторами процессов метаболизма (АМФ) и внутриклеточными посредниками действия гормонов (цАМФ, цГМФ). Образование нуклеотидов в организме происходит главным образом в процессе поэтапной сборки из эндогенных предшественников, реже путём реутилизации азотистых оснований, высвобождаемых при расщеплении нуклеиновых кислот в тканях. Скорость синтеза пуриновых и пиримидиновых рибо- и дезоксирибонуклеотидов в организме является объектом тонкой регулировки. Знание процессов биосинтеза и распада нуклеотидов позволяет понять патогенез ряда заболеваний, связанных с нарушениями обмена пуринов и пиримидинов (подагра, ксантинурия, оротацидурия), грамотно использовать в качестве противоопухолевых препаратов структурные аналоги азотистых оснований, нуклеозидов (6-меркаптопурин, 5-фторурацил, фторафур) и антагонисты коферментов, участвующих в биосинтезе нуклеотидов (аметоптерин, аминоптерин).
Цель занятия. После изучения данной темы студент должен знать особенности биосинтеза и распада пуриновых и пиримидиновых нуклеотидов, механизмы регуляции этих процессов и основные виды их нарушений, уметь применять приобретённые знания для решения теоретических и практических задач.
Исходный уровень знаний.
Строение пуриновых и пиримидиновых азотистых оснований, нуклеозидов, нуклеотидов, их роль в организме.
Строение аминокислот (глицин, глутамин, аспартат).
Пентозофосфатный путь окисления: роль в организме.
Коферментные функции витаминов (фолиевая кислота).
Механизмы регуляции активности ферментов.
Принципы диагностики врождённых дефектов ферментов.
5.1. Катаболизм нуклеопротеинов.
5.1.1. Нуклеопротеины – сложные белки, содержащие в качестве простетической группы нуклеиновые кислоты (РНК или ДНК). В зависимости от того, какая кислота входит в их состав, различают рибонуклеопротеины и дезоксирибонуклеопротеины.
Структурными единицами РНК и ДНК являются нуклеотиды, каждый из которых в свою очередь состоит из азотистого основания и пентозы (образующих нуклеозид), соединённых с остатком фосфорной кислоты.
Пентоза и азотистое основание в каждом нуклеотиде связаны N-гликозидной связью, фосфат и пентоза – сложноэфирной связью. Отдельные нуклеотиды в ДНК или РНК связаны 3’,5’-фосфодиэфирной связью (см. рисунок).
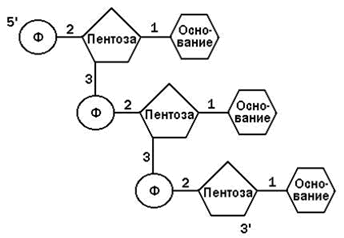
Рисунок 5.1. Схема строения фрагмента полинуклеотидной цепи. Примечание. Пентоза – рибоза в РНК, дезоксирибоза в ДНК. Азотистое основание – аденин, гуанин, цитозин, урацил в РНК, аденин, гуанин, цитозин, тимин в ДНК. Цифрами обозначены типы связей: 1 – N-гликозидные; 2 – сложноэфирные в нуклеотиде; 3 – 3’,5’-фосфодиэфирные (между нуклеотидами).
5.1.2. Катаболизм нуклеопротеинов в желудочно-кишечном тракте. Нуклеопротеины, поступающие в организм с пищей, подвергаются расщеплению под действием протеолитических ферментов кишечника. Высвобождаемые нуклеиновые кислоты гидролизуются при участии рибонуклеаз и дезоксирибонуклеаз панкреатического сока до мононуклеотидов. Действие панкреатических нуклеаз дополняют фосфоэстеразы кишечного сока. Схематично это можно представить следующим образом:
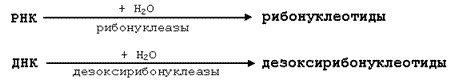
Далее, под воздействием нуклеотидаз и фосфатаз происходит гидролиз нуклеотидов до нуклеозидов, которые либо всасываются, либо под воздействием ферментов слизистой кишечника расщепляются до пуриновых и пиримидиновых оснований.
В организме человека большая часть пуриновых и пиримидиновых оснований, высвободившихся из нуклеиновых кислот, которые поступают с пищей, превращается в конечные продукты (при этом не происходит их включения во вновь образующиеся молекулы нуклеиновых кислот в тканях организма). То есть, нуклеиновые кислоты пищи практически не выступают в роли поставщика непосредственных предшественников нуклеиновых кислот тканей организма.
В то же время нуклеотиды и нуклеозиды, введённые парентерально, могут включаться в нуклеиновые кислоты без всяких изменений. Это послужило основой методов исследования метаболизма нуклеиновых кислот путём введения меченных радиоактивными изотопами азотистых оснований.
2.2. Распад пуриновых и пиримидиновых нуклеотидов до конечных продуктов в тканях.
Распад пуриновых нуклеотидов.
Аденозин и гуанозин, которые образуются при гидролизе пуриновых нуклеотидов, подвергаются ферментативному распаду с образованием конечного продукта – мочевой кислоты, которая выводится с мочой из организма.
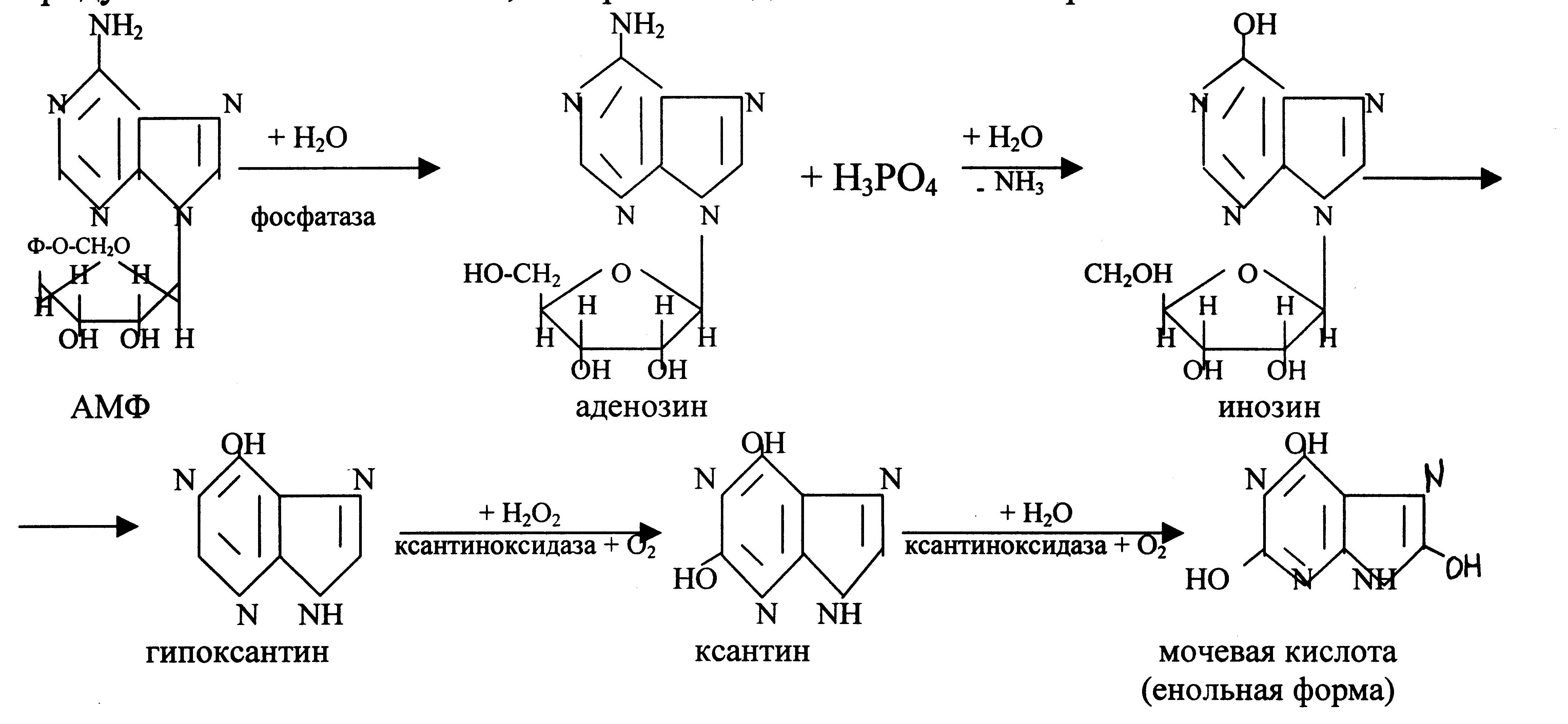
Распад пиримидиновых нуклеотидов.
Начальные этапы этого процесса катализируются специфическими ферментами. Конечные продукты: СО2, NН3, мочевина, β-аланин, β-аминоизомасляная кислота. β-аланин используется для синтеза дипептидов мышц – карнозина и ансерина или выделяется с мочой.

Биосинтез пуриновых, пиримидиновых нуклеотидов в тканях.
Биосинтез пуриновых мононуклеотидов.
Первоначальным соединением синтеза служит Д-рибозо-5-фосфат, который является продуктом пентозофосфатного цикла и на который переносится пирофосфатная группа АТФ. Образовавшийся 5-фосфорибозил-1-пирофосфат (ФРПФ) взаимодействует с глутамином, который является донором NН2-группы в результате чего образуется β-5-фосфорибозил-амин. Эта стадия становится ключевой в синтезе пуринов. Затем присоединяется молекула глицина к свободной NН2-группе β-5-фосфорибозил-амина с образованием глицинамидрибонуклеотида. Еще через несколько стадий образуется первый пуриновый нуклеотид инозинмонофосфат (ИМФ), из которого затем синтезируются остальные нуклеозидфосфаты.

Биосинтез пиримидиновых нуклеотидов
Первоначальными соединениями этого процесса являются карбамоилфосфат и аспарагиновая кислота. Из них через длинную цепь реакций образуется уридинмонофосфат (УМФ) и остальные пиримидиновые нуклеотиды.

2.4. Заболевания, связанные с нарушением обмена нуклеотидов: подагра, синдром Леша-Нихена.
Гиперурикемия – повышение в плазме крови концентрации мочевой кислоты. Вследствие гиперурикемии может развиться подагра.
Подагра – заболевание, вызванное нарушением обмена нуклеиновых кислот. В хрящах, сухожилиях, в суставных сумках, иногда в почках, коже, мышцах откладываются кристаллы мочевой кислоты и уратов. Вокруг этих отложений образуется воспаление и грануляционный вал, который окружает омертвевшую ткань, при этом образуются подагрические узлы — тофусы (в суставах пальцев рук, ног, в хрящах ушной раковины), что сопровождается деформацией и болезненностью пораженных суставов. К характерным признакам подагры относятся повторяющиеся приступы острого воспаления суставов (чаще всего мелких) – острого подагрического артрита. Обычно больные склонны к атеросклерозу и гипертонии. В их крови наблюдается большая концентрация мочевой кислоты – гиперурикемия. В течение нескольких дней перед приступом подагры увеличивается выделение воды и хлорида натрия с мочой, т.е. сдвигается водно-солевой баланс. Вследствие этого возрастает концентрация мочевой кислоты в крови и отложение ее в тканях. Как правило, подагра генетически детерминирована и носит семейный характер. Она вызвана нарушениями в работе фосфорибозилдифосфата (ФРДФ) синтетазы или гипоксантингуанин- или аденинфосфорибозилтрансфераз. К другим характерным проявлениям относят нефропатию, при которой наблюдают образование уратных камней в мочевыводящих путях.
Читайте также: Как вывести маркер с ткани в домашних условиях с одежды
Синдром Леша-Нихена – тяжелая форма гиперурикемии, которая наследуется как рецессивный признак, сцепленный с Х-хромосомой. Проявляется только у мальчиков. Кроме симптомов подагры наблюдаются церебральные параличи, нарушение интеллекта, попытки наносить себе раны (укусы губ, пальцев). Связана болезнь с дефектом фермента гипоксантин-гуанин-фосфорибозилтрансферазы, которая катализирует превращение гипоксантина и гуанина в гуанинимонофосфат (ГМФ), поэтому они превращаются в мочевую кислоту. В первые месяцы жизни неврологические расстройства не обнаруживаются, но на пеленках отмечают розовые пятна, вызванные присутствием в моче кристаллов мочевой кислоты. При отсутствии лечения больные погибают в возрасте до 10 лет из-за нарушения функции почек.
Основной препарат для лечения гиперурикемии – аллопуринол (структурный аналог гипоксантина).
Нарушения пуринового обмена
| Сайт: | Образовательный портал МБФ (ВолгГМУ) |
| Курс: | Избранные вопросы молекулярной патологии для клинических ординаторов 2020 |
| Книга: | Нарушения пуринового обмена |
Оглавление
1. Пуриновый обмен
Пуриновый обмен — совокупность процессов синтеза и распада пуриновых нуклеотидов. Пуриновые нуклеотиды состоят из остатка азотистого пуринового основания, углевода рибозы (дезоксирибозы), связанного бета-гликозидной связью с атомом азота пуринового основания, и одного или нескольких остатков фосфорной кислоты, присоединенных эфирной связью к атому углерода углеводного компонента.
Важнейшие азотистые основания
Мажорным пуриновым нуклеотидом является АТФ. В организме АТФ является одним из самых часто обновляемых веществ; так, у человека продолжительность жизни одной молекулы АТФ менее 1 мин. В течение суток одна молекула АТФ проходит в среднем 2000—3000 циклов ресинтеза (человеческий организм синтезирует около 40 кг АТФ в день, но содержит в каждый конкретный момент примерно 250 г), то есть запаса АТФ в организме практически не создаётся, и для нормальной жизнедеятельности необходимо постоянно синтезировать новые молекулы АТФ.
Аденозинтрифосфа́т или Аденозинтрифосфорная кислота (сокр. АТФ, англ. АТР) — нуклеозидтрифосфат, имеющий большое значение в обмене энергии и веществ в организмах. АТФ — универсальный источник энергии для всех биохимических процессов, протекающих в живых системах. Открытие вещества произошло в 1929 году группой учёных Гарвардской медицинской школы — Карлом Ломаном, Сайрусом Фиске и Йеллапрагадой Суббарао, а в 1941 году Фриц Липман показал, что АТФ является основным переносчиком энергии в клетке.

У человека и приматов мочевая кислота — конечный продукт обмена пуринов, образующийся в результате ферментативного окисления ксантина под действием ксантиноксидазы; у остальных млекопитающих мочевая кислота превращается в аллантоин. Небольшие количества мочевой кислоты содержатся в тканях (мозг, печень, кровь), а также в моче и поте млекопитающих и человека. При некоторых нарушениях обмена веществ происходит накопление мочевой кислоты и её кислых солей (уратов) в организме (камни в почках и мочевом пузыре, подагрические отложения, гиперурикемия). У птиц, ряда пресмыкающихся и большинства наземных насекомых мочевая кислота — конечный продукт не только пуринового, но и белкового обмена. Система биосинтеза мочевой кислоты (а не мочевины, как у большинства позвоночных) в качестве механизма связывания в организме более токсичного продукта азотистого обмена — аммиака — развилась у этих животных в связи с характерным для них ограниченным водным балансом (мочевая кислота выводится из организма с минимальным количеством воды или даже в твёрдом виде). Высохшие экскременты птиц (гуано) содержат до 25 % мочевой кислоты. Обнаружена она и в ряде растений.
1.1. Синтез пуринов de Novo
Основным путем образования пуриновых нуклеотидов является синтез из простых предшественников (de novo). В этом метаболическом пути свободное азотистое основание не образуется, а пуриновое кольцо формируется на остатке рибозо-5-фосфата при участии глицина, амидного азота Глн, а-NН2-группы Асп, СО2 и одноуглеродных производных: метенил- и формил-Н4-фолата. Синтез пуриновых оснований происходит во всех клетках организма , главным образом в печени. Исключение составляют эритроциты, полиморфноядерные лейкоциты, лимфоциты.
Условно все реакции синтеза можно разделить на 4 этапа:
1. Синтез 5′-фосфорибозиламина
Первая реакция синтеза пуринов заключается в активации углерода в положении С 1 рибозо-5-фосфата, это достигается синтезом 5-фосфорибозил-1-дифосфата (ФРДФ). Рибозо-5-фосфат является тем якорем, на основе которого синтезируется сложный пуриновый цикл.
Вторая реакция – это перенос NH 2-группы глутамина на активированный атом С 1 рибозо-5-фосфата с образованием 5′-фосфорибозиламина . Указанная NH 2-группа фосфорибозиламина уже принадлежит будущему пуриновому кольцу и ее азот будет атомом номер 9.
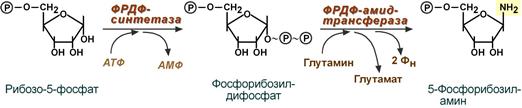
Реакции синтеза 5′-фосфорибозиламина
2. Синтез инозинмонофосфата
5-фосфорибозиламин вовлекается в девять реакций, и в результате образуется первый пуриновый нуклеотид – инозинмонофосфорная кислота (ИМФ). В этих реакциях источниками атомов пуринового кольца являются глицин, аспартат, еще одна молекула глутамина, углекислый газ и производные тетрагидрофолиевой кислоты (ТГФК). В целом на синтез пуринового кольца затрачивается энергия 6 молекул АТФ.
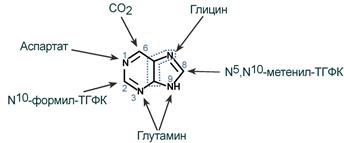
Источники атомов пуринового кольца
В схеме представлена последовательность всех 11 химических реакций этого синтеза с указанием ферментных систем, коферментов, источников энергии и других известных к настоящему времени кофакторов.

3. Синтез аденозинмонофосфата и гуанозинмонофосфата
- Гуанозинмонофосфат (ГМФ) образуется в двух реакциях – сначала он окисляется ИМФ-дегидрогеназой до ксантозилмонофосфата, источником кислорода является вода, акцептором водорода – НАД. После этого работает ГМФ-синтетаза, она использует универсальный клеточный донор NH 2-групп – глутамин, источником энергии для реакции служит АТФ.
- Аденозинмонофосфат (АМФ) также образуется в двух реакциях, но в качестве донора NH 2-группы выступает аспарагиновая кислота. В первой, аденилосукцинат-синтетазной, реакции на присоединение аспартата используется энергия распада ГТФ, во второй реакции аденилосукцинат-лиаза производит удаление части аспарагиновой кислоты в виде фумарата.
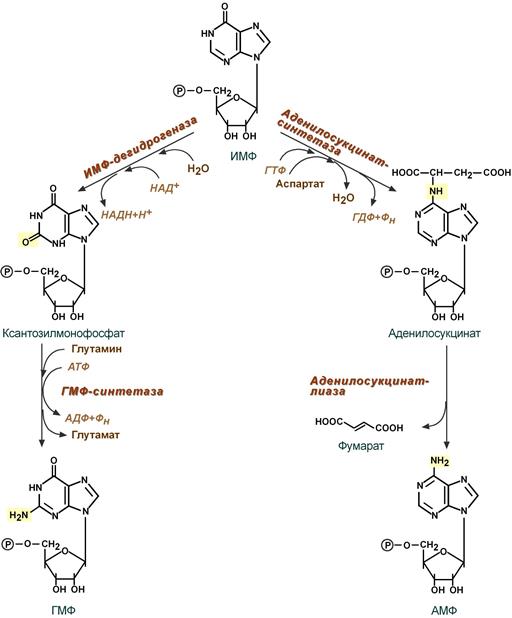
4. Образование нуклеозидтрифосфатов АТФ и ГТФ.
Синтез ГТФ осуществляется в 2 стадии посредством переноса макроэргических фосфатных групп от АТФ. Синтез АТФ происходит несколько иначе. АДФ из АМФ образуется также за счет макроэргических связей АТФ. Для синтеза же АТФ из АДФ в митохондриях есть фермент АТФ-синтаза , образующий АТФ в реакциях окислительного фосфорилирования.
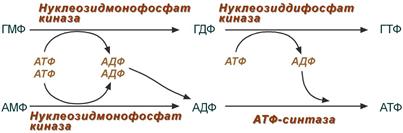
Другим минорным источником АТФ является субстратное фосфорилирование в гликолизе и ЦТК
Образование дезоксирибонуклеотидов
Особенностью обмена пуринов является то, что они могут образовывать не только рибонуклеотиды, но и дезоксирибонуклеотиды.
Читайте также: Как изготовить ткань из пряжи
Дезоксирибонуклеозидтрифосфаты необходимы клетке для синтеза ДНК . Их образование протекает в три реакции, первая и третья реакции просты и понятны. Главные события происходят во второй реакции.
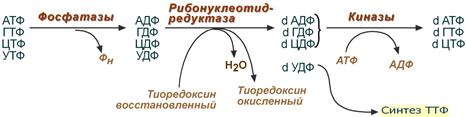
Все три реакции синтеза дезоксирибонуклеотидов
1. Реакция дефосфорилирования
В самом начале процесса происходит потеря рибонуклеозидтрифосфатами одной фосфатной группы и образуются АДФ, ГДФ, ЦДФ, УДФ.
2. Реакция восстановления
Во второй реакции фермент рибонуклеозид-редуктаза восстанавливает АДФ, ГДФ, ЦДФ, УДФ до дезоксирибонуклеозиддифосфатов dАДФ, dГДФ, dЦДФ, dУДФ. Донором водорода для восстановления рибозы является белок тиоредоксин , его SH-группы окисляются кислородом рибозы и образуется вода. Последующее восстановление тиоредоксина в рабочее состояние обеспечивается за счет НАДФН.

Механизм реакции синтеза дезоксирибонуклеотида
3. Реакция фосфорилирования
После образования dАДФ, dГДФ, dЦДФ фосфорилируются, а dУДФ используется для синтеза тимидилового нуклеотида.
Тиоредоксины
Тиоредоксины — семейство маленьких белков, представленный во всех организмах от архей до человека. Они участвуют во многих важных биологических процессах, включая определение окислительно-восстановительного потенциала клетки и передачу сигнала. У человека тиоредоксин кодируется геном TXN. Мутации, приводящие к потери функциональности даже одного аллеля этого гена, приводят к смерти на стадии четырёхклеточного эмбриона. Тиоредоксин играет значительную роль в организме человека, хотя и не до конца ясно какую именно. Всё чаще и чаще его возможные функции связывают с действием лекарств и противодействием активным формам кислорода. У растений тиоредоксины регулируют целый спектр жизненно важных функций, начиная от фотосинтеза и роста и заканчивая цветением, развитием и прорастанием семян. А совсем недавно выяснилось, что они также участвует в межклеточном взаимодействии и обмене информацией между растительными клетками
Тиоредоксины представляют собой белки с массой около 12 кДа. Их отличительная особенность — наличие двух расположенных рядом остатков остатков цистеина, заключённых в мотив типа CXXC, где С — цистеин, а Х — любая, как правило гидрофобная, аминокислота. Ещё одна отличительной черта всех тиоредоксинов — специфическая третичная структура, которая называется тиоредоксиновой укладкой.
Главной частью белка является дисульфидная связь. При помощи этой неё он может восстанавливать дисульфидные связи других белков, разрушая в них дисульфидные мостики. Таким образом он регулирует активность некоторых ферментов. Кроме того, восстанавливая дисульфидные связи, тиоредоксин поставляет электроны, которые затем используются во многих биохимических процессах клетки. Например, вместе с глутатионом он поставляет электроны для рибонуклеотидредуктазы, то есть участвует в синтезе дезоксинуктлеотидов, и ФАФС-редуктазы. В этом плане, его функция сходна с таковой у глутатиона и частично с ней перекрывается. Так, тиоредоксин является сильным антиоксидантом: вместе с глутатионовой системой тиоредоксиновая система участвует в обезвреживание активных форм кислорода, передовая электроны различным пероксидазам. Исследования показали, что тиоредоксин взаимодействует с рибонуклеазой, хориогонадотропинами, факторами коагуляции, глюкокортикоидным рецептором и инсулином. Реакцию тиоредоксина с инсулином традиционно используют для определения активности тиоредоксина. Было показано, что тиоредоксин способен стимулировать связывание факторов транскрипции с ДНК. Эти факторы были определены как ядерный фактор NF-κB, который является важным фактором в клеточной реакции на окислительный стресс, апоптоз и процессы опухолеобразования.
Восстановление тиоредоксина осуществляет специальный флавопротеин тиоредоксин редуктаза, который использует для этого одну молекулу НАДФН. Глутаредоксины во многом сходны по функциям с тиоредоксинами, но вместо специфической редуктазы они восстанавливаются глутатионом.
| ↔ 2 H + + 2 e — + | ||
| Восстановленный тиоредоксин | Окисленный тиоредоксин |
Способность тиоредоксинов противостоять окислительному стрессу была продемонстрирована в эксперименте с трансгенными мышами у которых была повышенная экспрессия тиоредоксина. Трансгенные мыши лучше сопротивлялись воспалительным реакциям и жили на 35 % дольше. Такие данные служат существенным аргументом в пользу свободнорадикальной теории старения. Тем не менее, результаты исследования нельзя считать достоверными, поскольку контрольная группа мышей жила значительно меньше обычного, что могло создать иллюзию увеличения продолжительности жизни у трансгенных мышей [9] .
У растений существует очень сложная система тиоредоксинов, состоящая из шести хорошо различимых типов (тиоредоксины f, m, x, y, h, и o). Они расположены в разных частях клетки и участвуют в массе различных процессов. Именно действие тиоредоксинов лежит в основе светозависимой активации ферментов. На свету, в результате совместного действия фотосистемы I и фотосистемы II образуется большое количество восстановительных эквивалентов — ферредоксинов. По достижении определённой концентрации ферредоксина, за счёт действия Фермент ферредоксин-тиоредоксинредуктазы происходит восстановление тиоредоксина, который в свою очередь активирует ферменты, восстанавливая дисульфидные связи. Таким путём активируется по крайней мере пять ключевых ферментов цикла Кальвина, а также белок-активаза Рубиско, альтернативная оксидаза митохондрий и терминальная оксидаза хлоропластов. Механизм активации через тиоредоксин позволяет регулировать активность ферментов не только в зависимости от соотношения НАДФН/НАДФ + , но и одновременно от интенсивности света. В 2010 году была открыта необычная способность тиоредоксинов перемещаться из клетки в клетку. Такая способность лежит в основе нового, ранее не известного для растений, способа межклеточной коммуникации
1.2. Переваривание нуклеиновых кислот и резервные источники пуриновых оснований

«Запасные» пути синтеза пуриновых нуклеотидов (реутилизация азотистых оснований и нуклеозидов)
Огромные затраты энергии для синтеза пуриновых нуклеотидов de novo не способны полностью обеспечить субстратами синтез нуклеиновых кислот в период гаструляции и раннего роста ребёнка. Потребность в большом количестве нуклеотидов привела к развитию «запасных» путей синтеза этих «дорогих» молекул. Наибольшее значение в этом процессе имеют ферменты, осуществляющие превращение пуринов в мононуклеотиды с использованием ФРДФ как донора остатка фосфорибозы.
Синтез АМФ и ГМФ из аденина и гуанина
ФРДФ-зависимое фосфорибозилирование пуринов катализируют 2 фермента.
- Аденинфосфорибозилтрансфераза (APRT), ответственная за образование АМФ (рис. 10-6).
- Гипоксантин-гуанинфосфорибозилтрансфераза (HGPRT), катализирующая образование ИМФ и ГМФ из гипоксантина и гуанина соответственно.
Однако в организме при любых ситуациях этот путь синтеза пуриновых нуклеотидов, получивший название «Salvage pathways», имеет вспомогательное значение.
Нуклеозидкиназы
Нуклеозиды, получающиеся при катаболизме нуклеиновых кислот из нуклеотидов под действием нуклеотидаз, могут повторно фосфорилироваться, образуя нуклеозид-5′-монофосфаты за счёт переноса γ-фосфатного остатка АТФ на соответствующий субстрат. У млекопитающих такой путь пополнения запасов пуриновых нуклеотидов в клетке не имеет существенного значения. Основным ферментом этой группы является аденозинкиназа, которая ускоряет реакцию:
Читайте также: Костный панцирь черепахи очень прочен живая костная ткань
Из всех способов реутилизации пуринов наиболее активна гипоксантин-гуанинфосфорибозилтрансферазная реакция, поскольку ИМФ, образующийся в этой реакции, вовлекается в синтез АМФ и ГМФ. Использование гипоксантина и гуанина по запасному пути становится жизненно важным событием в клетках, не способных к синтезу пуриновых нуклеотидов de novo. Значение аденинфосфорибозилтрансферазы в повторном использовании аденина менее существенно. По сравнению с аденозином количество аденина в клетках мало, а первый возвращается в фонд нуклеотидов с помощью аденозинкиназы.

Рис. Фосфорибозилирование аденина в АМФ.

Рис. Фосфорибозилирование гипоксантина и гуанина с образованием ИМФ и ГМФ

Синтез AMP из IMP и сохранение IMP через катаболизм AMP имеют чистый эффект дезаминирования аспартата на фумарат. Этот процесс был назван пуриновым нуклеотидным циклом (см. Диаграмму ниже). Этот цикл очень важен в мышечных клетках. Увеличение активности мышц создает потребность в увеличении цикла TCA, чтобы генерировать больше NADH для производства ATP. Однако у мускула не хватает большинства ферментов основных анаплевротических реакций. Мышцы пополняют промежуточные продукты цикла TCA в форме фумарата, продуцируемого пуриновым нуклеотидным циклом.

Пуриновый нуклеотидный цикл служит важной функцией при тренировке мышц. Генерация фумарата обеспечивает скелетную мышцу своим единственным источником анаплеротического субстрата для цикла TCA. Для продолжения работы цикла во время тренировки мышечный белок должен быть использован для доставки аминокислот для образования аспартата. Генерация аспартата происходит по стандартным реакциям трансаминации, которые взаимопревращают аминокислоты с α-кетоглутаратом с образованием глутамата и глутамата с оксалацетатом с образованием аспартата. Myoadenylate deaminase — это мускуло-специфическая изоформа АМФ-дезаминазы, а недостатки в myoadenylate deaminase приводят к усталости после тренировки, судорогам и миалгии.
1.3. Регуляция пуринового обмена
Следует отдельно подчеркнуть существование в клетках весьма тонкого механизма регуляции синтеза пуриновых нуклеотидов . Синтез их тормозится конечными продуктами по принципу обратной связи, т.е. ингибированием первой стадии переноса аминогруппы глутамина на ФРПФ. Фермент , катализирующий эту стадию, оказался аллостерическим регуляторным ферментом . Вторая особенность механизма регуляции заключается в том, что избыток ГМФ в клетках оказывает аллостерическое торможение только на свой собственный синтез, не влияя на синтез АМФ , и, наоборот, накопление АМФ подавляет свой синтез, не ингибируя синтеза ГМФ.
2. Нарушения пуринового обмена
К наиболее важным нарушениям пуринового обмена относятся избыточное образование и накопление мочевой кислоты, например при подагре и синдроме Леша — Найхана. В основе последнего лежит наследственная недостаточность фермента гипоксантинфосфатидилтрансферазы, вследствие чего свободные пурины не используются повторно, а окисляются в мочевую кислоту.

Клинические проблемы, связанные с метаболизмом нуклеотидов у людей, в основном являются результатом аномального катаболизма пуринов. Клинические последствия аномального метаболизма пуринов варьируются от умеренных до тяжелых и даже фатальных расстройств. Клинические проявления аномального катаболизма пуринов возникают из-за нерастворимости побочного продукта деградации, мочевой кислоты. Подагра является условием, которое возникает в результате осаждения урата в виде кристаллов мононатриевого урата (MSU) или дигидрата пирофосфата кальция (CPPD) в синовиальной жидкости суставов, что приводит к сильному воспалению и артриту. Воспалительный ответ связан с наличием кристаллов, вовлеченных в каспазу-1-активирующее воспаление, что приводит к получению интерлейкина-1β (IL-1β) и IL-18. Большинство форм подагры являются результатом избыточного производства пуринов и последующего катаболизма или частичного дефицита фермента спасения, HGPRT. Большинство форм подагры можно лечить, вводя антиметаболит: аллопуринол. Это соединение является структурным аналогом гипоксантина, который сильно ингибирует ксантиноксидазу.
Два серьезных нарушения, оба достаточно хорошо описаны, связаны с недостатками в пуриновом обмене: синдром Леша-Нихана и тяжелое комбинированное иммунодефицитное заболевание (SCID). Синдром Леша-Нихана является результатом потери функционального гена HGPRT. Расстройство наследуется как связанная с полом черта с геном HGPRT на Х-хромосоме (Xq26-q27.2). Пациенты с этим дефектом проявляют не только тяжелые симптомы подагры, но также серьезную неисправность нервной системы. В наиболее серьезных случаях пациенты прибегают к саморазрушению. Смерть обычно возникает прежде, чем пациенты достигнут двадцатилетия.
SCID относится к группе потенциально смертельных расстройств из-за комбинированной потери функции как T-, так и B-лимфоцитов. Существует, по меньшей мере, 13 известных и охарактеризованных генетических причин SCID. Наиболее распространенной (45%) причиной SCID является Х-связанное расстройство, вызванное потерей функции общей гамма-γ-цепи рецептора Т-клеток и других рецепторов интерлейкина (IL). Вторая наиболее распространенная (15%) форма SCID вызвана дефектами фермента аденозиндезаминазы, ADA. Это фермент, ответственный за превращение аденозина в инозин в катаболизме пуринов. Этот дефицит избирательно приводит к разрушению B и T лимфоцитов, клеток, которые создают иммунные ответы. В отсутствие ADA дезоксиаденозин фосфорилируется с образованием уровней dATP, которые в 50 раз выше, чем обычно. Уровни особенно высоки в лимфоцитах, которые имеют обильное количество спасительных ферментов, включая нуклеозид-киназы. Высокие концентрации dATP ингибируют рибонуклеотидредуктазу (см. Ниже), тем самым предотвращая образование других dNTP. Чистым эффектом является ингибирование синтеза ДНК. Поскольку лимфоциты должны быть способны резко размножаться в ответ на антигенный вызов, невозможность синтезировать ДНК серьезно ухудшает иммунные реакции и болезнь. Накопительный dATP также индуцирует разрывы ДНК-цепей в не делящихся лимфоцитах посредством прямой активации основной протеазы ( каспаза 9), участвующих в апоптозе (запрограммированная гибель клеток). Кроме того, активность S-аденозиломоцистеинагидролазы заметно ингибируется 2′-дезоксиаденозином, что приводит к накоплению S-аденозилгомоцистеина, что, в свою очередь, приводит к уменьшению синтеза S-аденозилметионина (AdoMet), критического субстрата в реакциях трансметилирования. ADID с дефицитом SCID обычно смертелен в младенчестве, если не приняты специальные защитные меры. Менее тяжелый иммунодефицит возникает при отсутствии активности пуриновой нуклеозидфосфорилазы (PNP), другого фермента, разрушающего пурин.
Одна из многих заболеваний, связанных с сохранением гликогена, болезнь Гирке также приводит к чрезмерному производству мочевой кислоты. Это расстройство является следствием дефицита глюкозо-6-фосфатазной активности. Повышенная доступность глюкозо-6-фосфата увеличивает скорость потока через пентозо-фосфатный путь, что приводит к повышению уровня рибозо-5-фосфата и, следовательно, ФРДФ. Увеличение ФРДФ приводит к избыточному биосинтезу пуринов с последующим катаболизмом к мочевой кислоте.
- Свежие записи
- Балкон в многоквартирном доме: является ли он общедомовым имуществом?
- Штраф за остекление балкона в 2022: что это и как избежать наказания
- Штраф за мусор с балкона: сколько заплатить за выбрасывание окурков
- Оформление балконного окна: выбираем шторы из органзы
- Как выбрать идеальные шторы для маленькой кухни с балконом
- Правообладателям
- Политика конфиденциальности