
Оценка носовой кости в I триместре беременности: как, где, когда и зачем мы делаем

УЗИ сканер HS60
Профессиональные диагностические инструменты. Оценка эластичности тканей, расширенные возможности 3D/4D/5D сканирования, классификатор BI-RADS, опции для экспертных кардиологических исследований.
Задача пренатального скрининга — выявление беременных женщин группы высокого риска по рождению детей с хромосомными болезнями и врожденными пороками развития с целью более детального анализа состояния плода с помощью специальных методов [1].
С конца прошлого века в алгоритм пренатального скрининга включен расчет индивидуального комбинированного риска, центральное место в котором занимают ультразвуковой и биохимический скрининг в I триместре (11-14 нед беременности). Были созданы компьютерные программы расчета риска, учитывающие возраст, ультразвуковой маркер I триместра (толщина воротникового пространства — ТВП) и биохимические маркеры крови (β-hCG и PAPP-A) беременной женщины [2].
За последние 10 лет данная система полностью оправдала себя и получила дальнейшее развитие, путем прибавления к расчету риска добавочных ультразвуковых маркеров (оценка носовой кости, венозного протока, трикуспидальной регургитации, некоторых маркерных врожденных пороков развития). Расширение протокола осмотра с оценкой новых ультразвуковых маркеров (оценка носовой кости, кровоток в венозном протоке и на трикуспидальном клапане) улучшает чувствительность комбинированного скрининга благодаря увеличению частоты обнаружения и уменьшению частоты ложноположительных результатов [3].
Однако их оценка требует соответствующего углубленного обучения врача УЗД и получение сертификата компетентности на проведение данного вида исследования, так как только после получения доступа на конкретный вид исследования программа расчета риска будет учитывать эти данные в своих расчетах 3.
Преимуществами проведения УЗИ в 11-14 нед помимо установки точного срока беременности являются: ранняя диагностика многих пороков развития плода, оценка маркеров хромосомных аномалий для выявления беременных высокого риска по хромосомным аномалиям у плода, при многоплодной беременности именно в ранний срок возможно установить хориальность, что является важнейшим фактором, определяющим исход многоплодной беременности, возможность выявить женщин группы высокого риска по развитию преэклампсии в поздние сроки беременности [3, 4].
Копчико-теменной размер плода (КТР) для проведения скрининга I триместра должен быть в пределах 45-84 мм. Для оценки носовой кости в I триместре беременности необходимо соблюдать строгие условия. Это адекватное увеличение (на снимке должны быть только голова и верхняя часть грудной клетки), среднесагиттальный скан (должны быть визуализированы эхогенный кончик носа, небный отросток верхней челюсти, диэнцефалон), нос представлен тремя «К» (кончик носа, кожа, кость). Кожные покровы и кости носа визуализируются в виде знака «равенства», нос параллелен датчику.
Такие правила, как размер плода, адекватное увеличение, среднесагиттальный скан идентичны таковым при измерении ТВП. Таким образом, при выведении корректного скана для измерения ТВП, что является обязательным при проведении УЗ-исследования в сроки 11-14 нед беременности, оценка носовой кости проводится в том же самом срезе, не требуя получения дополнительных изображений.
Если все критерии соблюдены, то на уровне носа плода должны быть видны три четко различимые линии: верхняя линия представляет собой кожу, книзу от нее визуализируется более толстая и более эхогенная, чем кожа носовая кость. Третья линия, визуализируемая кпереди от носовой кости и на более высоком уровне, чем кожа — это кончик носа (рис. 1).
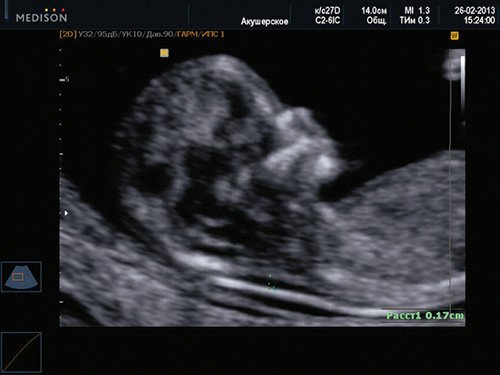
Рис. 1. Нормальная носовая кость.
Считается, что носовая кость нормальна, когда она по своей структуре более эхогенна, чем надлежащая кожа и патологична, если она не видна (аплазия) (рис. 2) или ее длина меньше нормы (гипоплазия) (рис. 3). В случае одинаковой или меньшей эхогенности носовой кости чем кожи носовая кость считается патологической (рис. 4).
а) Стрелкой указана эхогенная кожа плода.
б) Стрелкой указано отсутствие носовой кости.
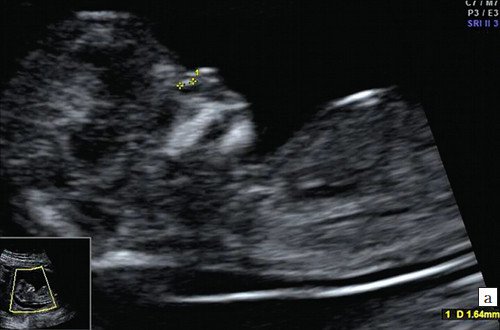
а) Носовая кость в 12 нед и 2 дня длиной 1,4 мм (меньше нижней границы нормы).
б) Носовая кость 2,1 мм в 14 нед у плода с синдромом Дауна.
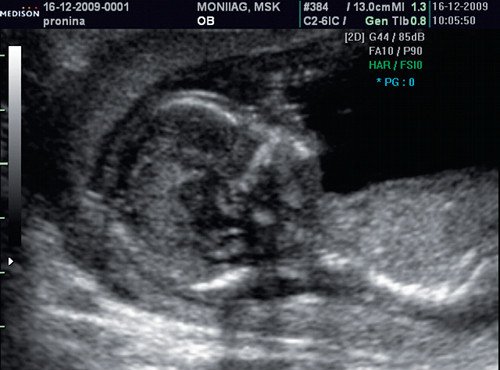
Рис. 4. Сниженная эхогенность носовой кости.
Итак, патологией носовой кости считается:
- отсутствие носовой кости (аплазия);
- изменение ее длины (гипоплазия);
- изменение ее эхогенности.
Учитывая то, что многие работы по изучению этого важного маркера были проведены на различных по составу группах населения, данные по частоте отсутствия носовой кости у разных авторов отличаются. Так, по усредненным данным по мультицентровым исследованиям FMF в 11-14 нед носовая кость отсутствует у эуплоидов (в случае нормального кариотипа) от 1 до 2,6% плодов [2, 5, 6], при хромосомных патологиях: у плодов с трисомией 21 — в 60%, с трисомией 18 — в 50%, у плодов с трисомией 13 — у 40% [3].
Проводились многочисленные работы, посвященные измерению и оценке носовой кости в срок 11-14 нед беременности. Некоторые авторы предлагают оценивать лишь ее наличие или отсутствие (+/-) [7]. Некоторые работы кроме оценки носовой кости посвящены ее измерению, сравнивая длину с нормативными для данного срока значениями 10.
Эволюция развития оценки этого маркера и мнение специалистов на этот счет, пожалуй, одна из самых дискутабельных проблем, не до конца решенных в скрининге I триместра беременности. Большинство авторов считают оценку носовой кости в I триместре одной из самых сложных задач среди всех остальных маркеров. И это мнение не лишено оснований.
Безусловно, сторонники теории о том, что для каждой расы (азиаты, афро-американцы и т.д.) и популяции народов (буряты, калмыки, народы Северного Кавказа) должны существовать свои процентильные нормативы для каждого КТР правы. Однако проведение этих исследований возможно лишь тогда, когда в рамках безвыборочного скрининга на нормальных плодах будут проведены мультицентровые исследования с измерением носовой кости.
В программе расчета риска Astraia при оценке носовой кости есть 4 поля: норма, патология (аплазия/гипоплазия), четко не видна, оценить не удалось, т.е. для того, чтобы поставить диагноз «Гипоплазия носовой кости» нужно удостовериться, что она на самом деле меньше нормативных значений для данного срока беременности, а это можно сделать только путем ее измерения и сравнения с известным нормативом.
Читайте также: Протеогликановые агрегаты в хрящевой ткани выполняют следующие функции
Метод оценки носовой кости только лишь «да/нет», когда предлагается только увидеть носовую кость и сравнить ее эхогенность с кожей весьма «аппаратозависим», т.е. очень вариабелен и зависит от технических настроек ультразвукового сканера. При получении «жесткого» изображения, характерного для некоторых ультразвуковых аппаратов со специфическими заводскими пресетами (настройками) для осмотра плода в I триместре, всегда эхогенность кожи будет сопоставима, т. е. одинакова с эхогенностью носовой кости. Таким образом, у врачей практического звена, не имеющих возможности работать на сканерах премиум класса, возникают объективные трудности с оценкой этого важного дополнительного диагностического маркера.
Как сторонники метода измерения носовой кости в 11-14 нед приведем данные по Московской области. Область является разнородной по населяющему ее национальному составу. В своей работе мы пользовались нормативными значениями длины носовой кости, опубликованными J. Sonek и соавт. в 2003 году [8], за нижнюю границу нормы принимая значение 5-го процентиля (таблица).
Экспертами окружных кабинетов Московской области проводилась оценка не только присутствия и отсутствия носовой кости, но и ее измерение у всех беременных женщин (около 150 тысяч обследованных за 3,5 года работы скрининга). Все 31 эксперт Московской области имеют действующий сертификат компетенции FMF как на ТВП, так и на оценку носовой кости. Проведенный анализ выявления патологии (аплазия/гипоплазия) носовой кости у плодов с хромосомной патологией показал, что из пренатально выявленных 266 случаев синдрома Дауна у плода в I триместре носовая кость была патологична в 248 случаях, что составляет 93,2%.
Это высокая частота патологии носовой кости при синдроме Дауна свидетельствует о правильно выбранном алгоритме оценки носовой кости от которого мы никогда не намерены отказываться, получая такие высокочувствительные результаты, особенно, что касается диагностики синдрома Дауна. В случаях выявления других хромасомных аномалий, частота выявления патологии носовой кости была сопоставима с данными литературы. При синдроме Эдвардса носовая кость патологична у 78 плодов, что составляет 71%, при синдроме Патау — у 24 (59%) плодов, при моносомии Х — в 24 (42%) случаях, при триплоидии — у 22 (49%) плодов.
Особо хотелось бы подчеркнуть, что в нашем исследовании было 10 беременных корейской национальности, попавших в группу риска по хромосомной патологии. У 4 из них была диагностирована патология носовой кости у плода. Можно было ожидать, что это этническая особенность, однако все данные плоды при пренатальном кариотипировании имели хромосомную патологию (трисомию 21). И, наоборот, у 6 плодов, имеющих нормальный кариотип как по длине, так и по эхогенности носовой кости были в пределах нормативных для данного срока значений.
В работах некоторых авторов установлено, что при трисомии 21 в I триместре беременности лишь у 25% плодов носовая кость отсутствовала, в более высокой частоте она была гипоплазирована (36%) [11].
Так как у нормальных плодов отсутствие носовой кости более характерно для срока 11 нед беременности, чем 13 нед, FMF дает практическую рекомендацию о том, что если в этот срок (11 — начало 12 нед) у плода отсутствует носовая кость при условии нормальных показателей других маркеров (ультразвуковых и биохимических) не стоит учитывать этот показатель при расчете индивидуального риска. В дальнейшем рекомендуется провести дополнительное ультразвуковое исследование через одну неделю. В том случае, если носовая кость останется патологична, необходимо учитывать этот факт при перерасчете величины индивидуального риска по хромосомным аномалиям [4].
Оценка носовой кости улучшает результаты комбинированного скрининга. Частота обнаружения патологии увеличивается с 90 до 93%. Частота ложноположительных результатов уменьшается с 3,0 до 2,5% [2, 3, 5, 6].
Таким образом, собственные данные позволяют нам рекомендовать оценивать носовую кость в сроки 11-14 нед по двум параметрам: эхогенность и длина, принимая за патологию носовой кости ее отсутствие, гипоплазию и снижение эхогенности.
Литература
- Баранов В.С., Кузнецова Т.В., Кащеева Т.К. и др. Современные алгоритмы и новые возможности пренатальной диагностики наследственных и врожденных заболеваний. Методические рекомендации. С.-Петербург, 2013. С. 23-46.
- Nicolaides K.H. Screening for fetal aneuploidies at 11-13 weeks//Prenatal diagnosis. 2011, 31: 7-15.
- Nicolaides K.H. Пер. с англ. Михайлова А., Некрасовой Е. Ультразвуковое исследование в 11-13+6 недель беременности. С.-Петербург, 2007. ИД «Петрополис», 142 с.
- http://www.fetalmedicine.org/the-11-13-weeks-scan
- Kagan K.O., Cicero S., Staboulidou I., Wright D., Nicolaides K.H. Fetal nasal bone in screening for trisomies 21, 18 and 13 and Turner syndrome at 11-13 weeks of gestation // Ultrasound Obstet Gynecol. 2009; 33: 259-264.
- Kagan K.O., Staboulidou I., Cruz J., Wright D., Nicoladides K.H. Two-stage first-trimester screening for trisomy 21 by ultrasound assessment and biochemical testing // Ultrasound Obstet Gynecol. 2010. V. 36. N 5. P. 542-547.
- Cicero S., Curcio P., Papageorghiou A., Sonek J., Nicolaides K. Absence of nasal bone in fetuses with trisomy 21 at 11-14 weeks of gestation: an observational study // Lancet 2001; 358:1665-1667.
- Sonek J.D., Mckenna D.,Webb D.,Croom C., Nicolaides K. Nasal bone length throughout gestation: normal ranges based on 3537 fetal ultrasound measurements // Ultrasound in Obstetrics & Gynecology. 2003. V. 21. N 2. P. 152-155.
- Kanellopoulos V., Katsetos C., Economides D.L. Examination of fetal nasal bone and repeatability of measurement in early pregnancy // Ultrasound Obstet Gynecol. 2003 Aug;22(2):131-4.
- Cicero S., Bindra R., Rembouskos G., Tripsanas C., Nicolaides K.H. Fetal nasal bone length in chromosomally normal and abnormal fetuses at 11-14 weeks of gestation // Matern Fetal Neonatal Med. 2002; 11: 400-402.
- Keeling J.W., Hansen B.F., Kjaer I. Pattern of malformations in the axial skeleton in human trisomy 21 fetuses // Am J Med Genet. 1997; 68: 466-471.
Читайте также: Поперечнополосатая мышечная ткань ядра
УЗИ сканер HS60
Профессиональные диагностические инструменты. Оценка эластичности тканей, расширенные возможности 3D/4D/5D сканирования, классификатор BI-RADS, опции для экспертных кардиологических исследований.
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма

УЗИ сканер HS50
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Синдром Тричера Коллинза (СТК) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
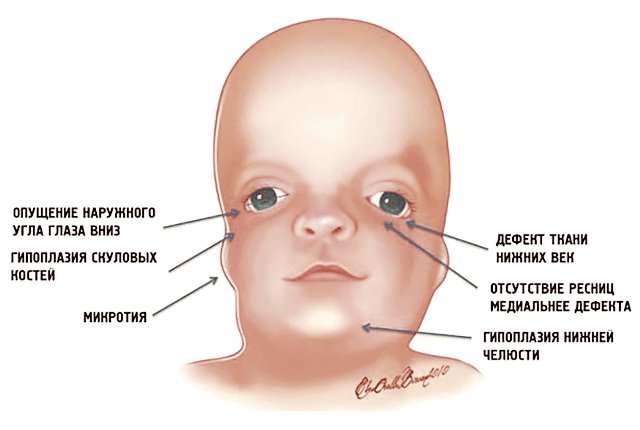
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Читайте также: Чем покрасить белую ткань в молочный цвет
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
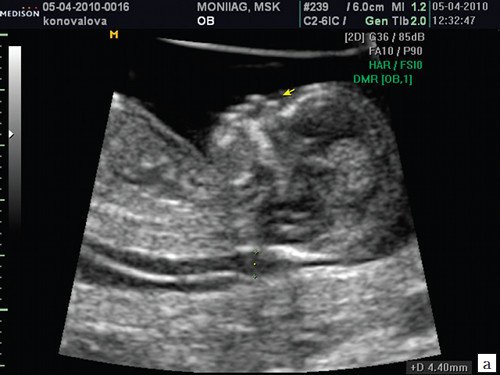
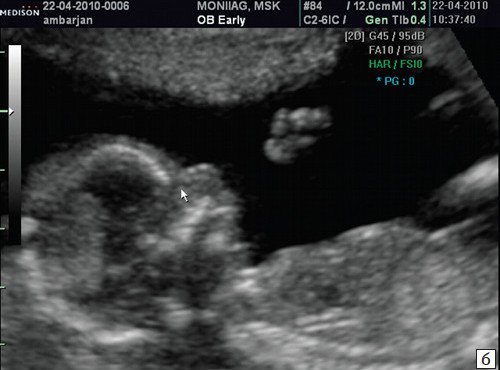
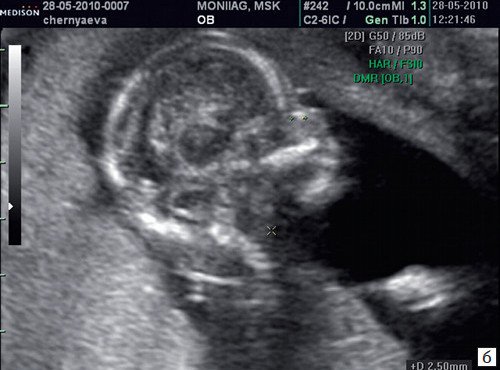
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
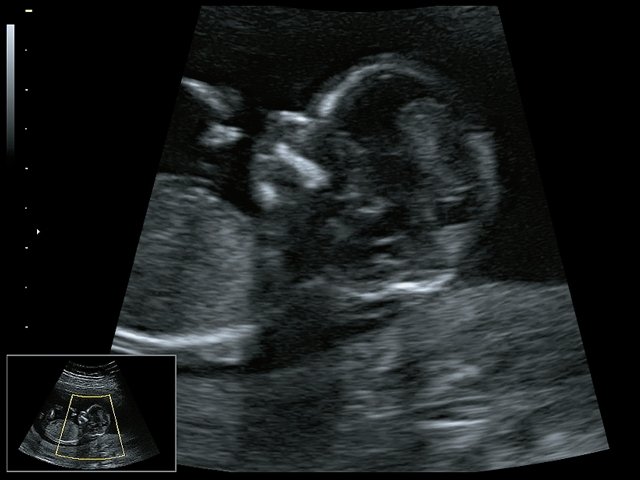
Рис. 2. Микрогнатия — сагиттальный скан в 2D, беременность 13 нед.
- Свежие записи
- Балкон в многоквартирном доме: является ли он общедомовым имуществом?
- Штраф за остекление балкона в 2022: что это и как избежать наказания
- Штраф за мусор с балкона: сколько заплатить за выбрасывание окурков
- Оформление балконного окна: выбираем шторы из органзы
- Как выбрать идеальные шторы для маленькой кухни с балконом